
Workflow involves mapping a reaction representation onto the activation energy of the stereocontrolling step
Scientists in Switzerland have developed a machine learning method that can determine the enantioselectivities of reactions catalysed by complex organocatalysts. Key to the strong performance of this machine learning technique is a clever trick to avoid time-consuming calculations, enabled by an informed choice of molecular descriptors, reaction representations and feature engineering.

Developing new catalysts is essential for ensuring quicker, more selective and more reliable reactions. ‘Experimentally, large-scale screenings remain expensive in terms of personnel resources, time and equipment needs,’ explains Simone Gallarati, from the Swiss Federal Institute of Technology Lausanne (EPFL), who is one of the lead researchers behind the study. ‘From a computational perspective, running computations on hundreds of catalytic systems is still a burdensome job, and achieving accurate predictions of enantioselectivity with standard methods is an incredibly difficult task.’ This is due to traditional computational methods needing to determine the transition states that lead to different enantiomers.
Cristina Trujillo, who was not involved in the study and researches the computational design of organocatalysts at Trinity College Dublin, Ireland, says calculating transition states in enantioselective reactions is usually very time-consuming and often sensitive to errors. ‘Small errors can lead to the predicted enantiomer being the opposite of that observed experimentally. In that sense, machine learning approaches, in general, provide an alternative solution to overcome the current challenges relating to computational cost.’
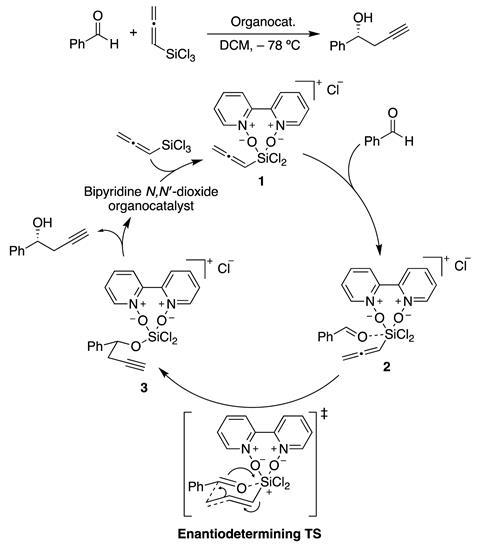
Gallarati and colleagues investigated if machine leaning methods could be used to determine the enantioselectivity, which arises from the relative activation energy of the (R)- and (S)-ligand configuration of the enantiodetermining transition states, of organocatalytic asymmetric propargylation which involves the reaction of an aldehyde with an allene and results in a new chiral centre. However, machine learning models are not without their complications. ‘In principle, we could feed a machine learning model information about an unknown catalyst – in the form of its 3D structure – and obtain within seconds a prediction of its selectivity,’ says Gallarati. ‘Unfortunately, a catalyst’s enantioselectivity is an incredibly difficult quantity to predict accurately with machine learning models.’
To overcome this challenge of predicting the enantioselectivity the team had to select an appropriate representation of the propargylation reaction and then fine-tune their model to sniff out the essential features from structural noise. This allowed for the machine learning algorithm to be trained to determine the activation energies for the competing (R) and (S) pathways that could then be translated to enantiomeric excess.
‘The fine ability of the presented strategy to predict energy differences is more than remarkable,’ comments Maria Besora, who researches catalysis using computational methods at the University of Rovira i Virgili in Spain.
Tailored reaction representations
Knowing that the cost of computing the enantiomeric transition states was challenging, the EPFL team explored using the intermediates either side of a transition state as the reaction representation to train a machine learning model. Starting with transition states from a database developed by Steven Wheeler and colleagues at Texas A&M University in the US, the EPFL team computed intermediates either side of the transition state using DFT intrinsic reaction coordinate calculations. These intermediates were then converted to molecular representations – a version of the important information about a molecule that can be understood by machine learning algorithms. Molecular representations ‘vary from collections of physical organic parameters, to text-based representations and chemoinformatics-type descriptors,’ says Gallarati. The team chose Slatm, which stands for Spectral London and Axilrod-Teller-Muto, as this representation can encode 3D molecular structures.
The next step involved finding a representation of the enantioselective reaction step that could be used for training and predicting activation energies. For this, the team explored the difference of the intermediate’s Slatm representations which ‘contains information on all the structural features that are changed during the reaction step, eliminating those that remain unchanged,’ according to Gallarati. This had the advantage of being a suitable representation of the reaction and reducing the amount of data the machine learning algorithm has to process. Finally, the team applied a feature engineering step involving cross-validation to improve accuracy and reduce the noise associated with the reaction representations, greatly decreasing the amount of data required.
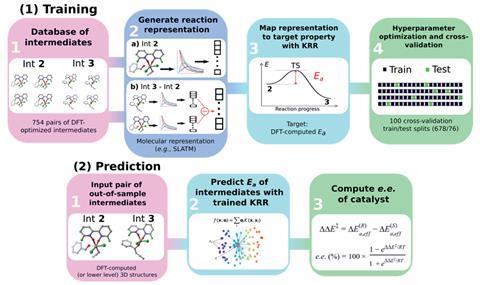
As a result, the machine learning model was able to predict the activation energy, and therefore the enantioselectivity, of bipyridine N,N’-dioxides, that were not part of the training database, only from intermediate structures. Furthermore, the machine learning model could elucidate the key features of the enantioselectivity determining transition states within the asymmetric propargylation reaction, identifying the presence of π-stacking and CH/π interactions as key motifs.
However, a limitation noted by Trujillo is that a high number of intermediates, over 1000, are required to train the algorithm and that the reaction investigated was quite specific. Yet, in the future it may be possible for machine learning solutions to be extended to a wider array of systems. ‘I think the use of machine learning models in the field of organocatalysis will increase in the near future. In this context, I do think it’s a promising development, but much time will be required for further generalisation,’ says Trujillo.
‘The fact that the strategy is not based on predicting enantiomeric excess but differences of energy opens the door to its applicability in other chemical problems, and also to prediction of enantioselectivity extended to the prediction of enantiomeric when more complex problems come into play,’ remarks Besora. ‘In principle, our approach can be used to develop a machine learning model to predict the enantioselectivity of any catalytic system,’ comments Gallarati, ‘provided a sufficiently large amount of structural information is available for training.’














No comments yet