
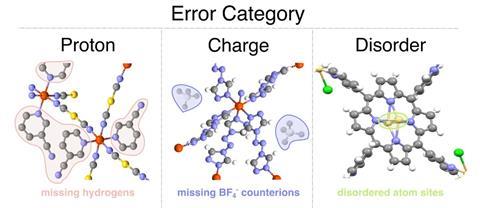
A neural network promises to improve the fidelity of crystal structure databases for metal–organic frameworks (MOF) by detecting and classifying structural errors.1 The approach, which flags entries with proton omissions, charge imbalances and crystallographic disorder, could help boost the accuracy of computational predictions used in materials discovery that rely on such databases.
Artificial intelligence and machine learning are becoming increasingly central to materials research, with scientists often turning to such tools to predict properties of new compounds. However, concerns are growing over the reliability of the underlying datasets; large crystal structure databases often contain errors that can compromise downstream simulations and predictions.
Marco Gibaldi, a PhD student in Tom Woo’s group at the University of Ottawa in Canada has experienced this issue firsthand with a MOF that was ranked as a top performer by predictive software. ‘When someone was trying to compare it against the experimental properties, the simulated properties and experiment were nothing alike, and we were trying to figure out what the issue was,’ says Gibaldi. They traced the discrepancy back to the simulated crystal structure, which contained errors in its structure and did not reflect a realistic chemical material.
Gibaldi and his colleagues have since analysed several open-access MOF databases commonly used for machine learning and found that more than 40% of the crystal structures in the databases contained errors.2
Now, Gibaldi, Woo and colleagues have developed a method to detect defects in crystal structures within databases and classify them according to their error type. They began by assembling a dataset of 11,000 MOFs, which required manually inspecting each structure and labelling errors by type, focusing on proton omissions, charge imbalances and crystallographic disorder. ‘It’s weirdly one of those things our group lovingly called “structure jail”, because a lot of us would be, for several months, just staring at structures and reading publications to try to figure out what the problem was,’ says Gibaldi.
The researchers then trained a graph attention neural network using the manually curated dataset to classify structural errors as either missing protons, charge imbalances or crystallographic disorder based on atomic number and oxidation state. When they tested the model on new datasets, including materials that aren’t MOFs, such as small molecules and transition metal complexes, it successfully classified up to 96% of proton and disorder errors, outperforming other feature-based classification models.
Yongchul Chung, a computational materials scientist at Pusan National University in South Korea, thinks the tool will mean researchers ‘do not waste their time on some of the structures that they found, which turn out to be incorrect.’
Beyond classifying errors, the work also serves as a timely reminder that machine learning models are only as reliable as the data they’re built on. ‘I think that’s something people should be aware of in a broader context,’ adds Chung. ‘It’s important not just to think about the model, but also the data it’s trained on. The work itself isn’t really new, but it’s one that’s worth continuing.’
Next, Woo’s team will focus on refining the graph representations to capture geometry more effectively and expanding the types of errors their model can detect. ‘[What] we’re trying to do is just move the needle a little closer so that those exchanges we have [between computational and experimental scientists] are a lot more rooted in the same language and they’re not just completely divorced from each other,’ Gibaldi says.
Chung agrees, noting that these tools will help build the trust in the computational research community and associated data sets.
References
1 M Gibaldi et al, J. Mater. Chem. A, 2025, 13, 32255 (DOI: 10.1039/d5ta05426e)
2 A J White et al, J. Am. Chem. Soc., 2025, 147, 21, 17579 (DOI: 10.1021/jacs.5c04914)












No comments yet