Platform computed thousands of stable designs for short proteins, up from two previous designs
The design and discovery of stable non-natural proteins, which could have far-reaching applications from medicines to materials, has entered a ‘new era’ thanks to a faster technique developed by researchers in the US and Canada. Combining computational design with advances in large-scale gene synthesis and high throughput screening, the team was able to identify thousands of synthetic miniproteins.
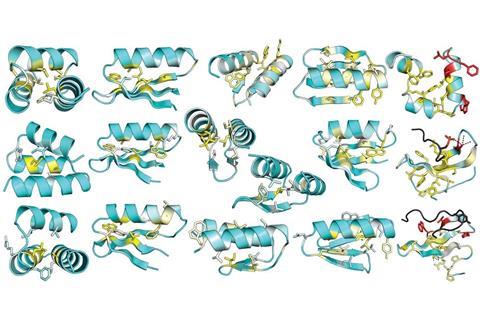
Miniproteins are made up of under 40 amino acid building blocks. By contrast, more complex proteins possess hundreds of amino acids. It’s the specific sequence of amino acids that determines how a protein folds into a stable 3D structure, which in turn governs how it functions. But understanding how the sequence directs folding has remained a puzzle.
For researchers wanting to design proteins with new functions not found in nature, knowing how sequence determines the stability of a fold is crucial in order to predict and find successful designs among the almost limitless possible amino acid sequences. Since miniproteins are less complex than larger proteins, they offer researchers a simpler way to understand the protein-folding problem.
However, previously only two stable miniproteins have been computationally designed. Part of the problem has been the cost of gene synthesis necessary for protein production – each designed protein requires its own custom synthesised piece of DNA. This has restricted studies to usually testing between just 10 to 20 designs at a time. Some larger studies have tested up to 100 protein designs but this still falls short when it comes to understanding why some designs are stable and others unravel.
Parallel platform
Now, David Baker’s lab at the University of Washington in Seattle, US, is leading the charge in developing a massively parallel protein design platform to create and test thousands of miniproteins. His team hopes to determine which designs work and why, thus enabling data analysis and further cycles to tweak and improve designs.
‘We avoid the cost of gene synthesis by having the genes for all the proteins synthesised together using high throughput OLS [oligo library synthesis] technology, and by using an assay where the proteins don’t need to be separated from each other to measure stability for each one,’ explains lead author Gabriel Rocklin in Baker’s Lab.
‘What is new and exciting about this approach is the scale: the proteins that they designed have been synthesised in parallel on a massive scale,’ comments Gail Bartlett, who investigates protein folding and design at the University of Bristol, UK. ‘This takes us a step closer towards data-driven protein design.’
OLS was originally developed for other laboratory protocols, such as large gene assembly, but Rocklin and his colleagues saw the potential for its use in up-scaling protein design.
Light assortment
The team tested the approach by designing 15,000 miniproteins and creating a way to spot which were stable. The researchers used yeast to express the miniprotein libraries, which involved each different yeast cell displaying many copies of one particular protein on its cell surface along with a fluorescent label. Following treatments with enzymes that can break down proteins, any miniproteins stable enough to survive this harsh treatment were collected, along with the cells they were hosted on, by fluorescence cell sorting and identified by deep sequencing. Four cycles of the design and screening process revealed 2788 stable protein structures that could have many bioengineering, pharmaceutical and synthetic biology applications.
‘Iterative analysis of design with experimental structural analysis is an important emerging principle in de novo protein design, powerfully enabled in this study by the large number of protein sequences for which experimental stability data could be generated,’ says Gaetano Montelione, who investigates protein structure at Rutgers University, US. ‘This is an outstanding advance.’
Rocklin is confident that industry and biotech could take advantage of the approach to find stable proteins. ‘Yeast display is already an important technique for selecting for proteins to bind different targets, and combining protease with yeast display could improve the stability of the selected proteins,’ he says. ‘In the longer term, our designed proteins could make an impact in industry as well. There aren’t many natural precedents for proteins this small and this stable, so these proteins are unique starting points for engineering new therapeutics.’
Birte Höcker, who studies protein design at the University of Bayreuth, Germany, agrees that the technique marks a ‘new era’ for protein design. ‘The robustness of predictability is what will make computational protein design become interesting for biotech and industry,’ she comments.
More complex designer proteins are yet to be demonstrated and OLS technology is limited in how long DNA fragments and hence the encoded proteins can be, but Rocklin suggests the approach should still work if large DNA libraries that encode larger proteins are used. Montelione agrees: ‘As DNA synthesis technologies continue to improve, I believe that this approach can be generalised to generate thousands of novel, larger proteins with a broad range of domain architectures.’
References
G J Rocklin et al, Science, 2017, 357, 168 (DOI: 10.1126/science.aan0693)












No comments yet