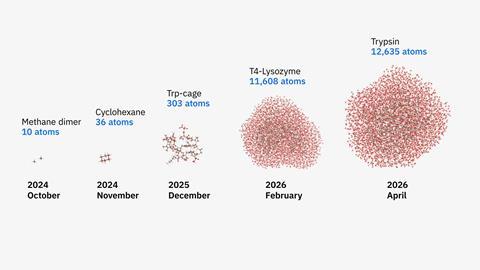
A new hybrid workflow involving two IBM quantum computers and two powerful supercomputers has allowed researchers to model two protein–ligand complexes containing up to 12,635 atoms – the largest simulations of biologically meaningful structures ever done with quantum hardware. The team from Cleveland Clinic in the US, Riken in Japan and IBM revealed their results in a preprint that has not been peer reviewed yet.

The researchers tested their method on two classic protein–ligand pairs: the digestive enzyme trypsin bound to the inhibitor benzamidine and T4 lysozyme bound to n-butyl-benzene, a model system used to study how small molecules bind inside proteins. ‘Quantum computers are now capable of tackling chemically and biologically relevant molecular systems,’ says Kenneth Merz from Cleveland Clinic and Michigan State University, US, who led the study. But this advancement didn’t come from quantum hardware alone. Instead, Merz and his colleagues split the task between different kinds of machines. First, classical supercomputers broke the large protein–ligand systems into smaller fragments. Then, IBM’s 156-qubit quantum processors calculated the quantum-mechanical behaviours of those pieces in tandem with two classical supercomputers. Finally, the results were put back together on classical systems to construct the full molecular picture.
What is a qubit?
Conventional computers run on binary logic – encoding information as 1s and 0s in various ways such as the orientation of magnetic poles in a computer’s memory chip. These 1s and 0s, or bits, can then be harnessed to perform computational calculations through the use of logic gates. These gates allow a current to flow when the gate’s logic rule is met.
A quantum bit, or qubit, is the quantum computing equivalent of a classical computing bit. Qubits still encode information as 1s or 0s, but encode them in states of a quantum object such as the up or down spin of an atom. However, while a classical bit can only be in one of two states (1 or 0), a qubit can be in a superposition of states, which effectively allows the two states to be mixed in any proportion. This means quantum computers should, in theory, be able to handle information more efficiently.
This potential has theoretical chemists excited because using a quantum object to model other quantum objects (such as atoms and electrons) should be much more efficient. Quantum computing therefore enables us to simulate molecules without having to make a compromise between accuracy and computational cost that is typically required when trying to simulate molecules with a classical computer. (For a more complete discussion of quantum computing examining the limitations and necessary simplications required to explain the concept we have a feature on the topic.)
‘This is a so-called quantum centric supercomputing model of computation (QCSC),’ explains Merz. ‘Rather than solving the entire problem at once, which is currently impossible both on quantum and classical hardware, we break down the problem into smaller, readily solvable sub-problems which can then be stitched back together again to give an accurate estimate of the answer you would get for the global problem if you could actually compute it. It’s the classic concept of dividing a problem so you can conquer it.’
Finding new drugs depends on understanding how molecules interact with proteins in the body. Scientists can use computers to predict that, but for the simulations to be useful, they need to be very accurate. Some of the current approaches are fast enough to handle large systems, but they rely on approximations that can miss important details. Others are much more accurate, but so computationally intensive that they become impractical for large biomolecules. As chemistry is governed by quantum mechanics, researchers believe that quantum computers may be especially well suited for tackling these problems that involve simulating the movement of a molecule’s electrons and their interactions with each other, being able to solve the underlying equations of chemistry more accurately than current methods while still keeping the calculations manageable.
‘This is a truly impressive paper,’ comments Lynn Kamerlin, a computational chemist and biochemist at Georgia Tech, US, who wasn’t involved in the research. ‘The authors have developed a new workflow for fragment-decomposition-based heterogeneous quantum–classical calculations with impressive accuracy compared to higher level computational approaches, significantly reducing computational cost,’ she says. ‘They have also effectively exploited quantum processors to realise those calculations and showcased the efficiency of these approaches to unprecedentedly large systems, breaking the 12,000-atom barrier.’ Kamerlin notes that protein simulations in classical computing are now pushing multi-million atoms. ‘But these calculations are significantly less expensive [computationally] than the quantum descriptions used in the current work,’ she adds.
The team also explicitly included a solvent for the first time in their QCSC calculations. They say that the systems they modelled in this way were about 40 times larger than those quantum computers could manage just six months ago – and that the accuracy of the simulations in a key step of the workflow also improved by up to 210 times.
‘It’s very hard to assess accuracy because the researchers use the performance of other computational approaches as a metric of accuracy,’ mentions Kamerlin. ‘I appreciate that they want to show how their approach performs compared to other more expensive computational approaches that would be likely more accurate but not tractable for these system sizes, but a comparison to experimental benchmarks is essential to be able to assess the utility of these results for drug discovery.’
Merz says that this approach can be used in many different fields. ‘We show two biological molecules with inhibitors bound, so structure-based drug design is one application area. However, this method can be applied to any molecular systems of interest in structural biology and materials science. It is a very flexible approach that can be applied to most chemical problems.’
References
K M Merz et al, 2026, arXiv, DOI: 10.48550/arXiv.2605.01138











No comments yet