
Biochemists in the US have found limits to what AlphaFold, the usually highly accurate protein structure prediction artificial intelligence system developed by Google DeepMind, can do. Lauren Porter’s team at the US National Library of Medicine at the National Institutes of Health looked at how AlphaFold performed with proteins that adopted more than one stable structure. The methods tested correctly identified one of seven proteins that could switch between structures that it hadn’t previously encountered.
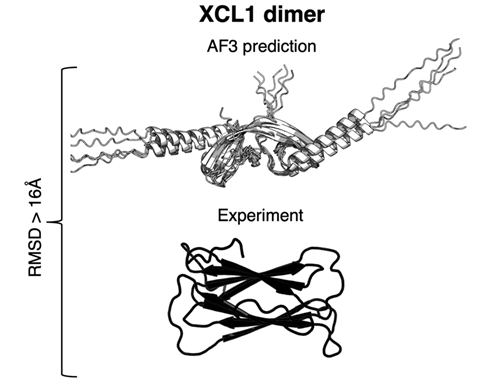
‘Current implementations of AlphaFold – the best protein structure predictor – are limited in their ability to predict fold-switching proteins,’ Porter tells Chemistry World. The study opens the ‘black box’ of how AlphaFold predicts a protein’s 3D structure from its amino acid sequence. The findings indicate that it’s memorising structures rather than considering how amino acids coevolve in related proteins, as scientists suggest happens in some methods. ‘If we want to use [AlphaFold] effectively, we need to understand what is driving these predictions,’ says team member Devlina Chakravarty.
In their natural environment, some proteins can remodel their structures in response to events in cells around them, Porter explains. That’s important in biological processes and diseases. Porter’s team therefore tries to measure, predict and understand such fold switching, even though it’s difficult.
AlphaFold is trained on the Protein Data Bank, which contains over 200,000 crystal structures. DeepMind hasn’t released AlphaFold’s training set, but the similar OpenFold model uses over 130,000 proteins. Very few of these adopt more than one structure. Porter explains that this makes fold-switching proteins ‘ideal’ to explore how AlphaFold works, as it hasn’t yet fully recognised patterns in their structures.
Porter’s team tested how often AlphaFold correctly predicted experimentally measured conformations of fold-switching proteins. The researchers considered identifying two distinct protein conformations a successful test. They tried every version of AlphaFold2, released in July 2021, and the AlphaFold3 server, released in May 2024. Her team also used two methods that the developers suggest reflects information on how amino acid sequences of fold-switching proteins coevolve.
Across the entire study the researchers generated more than 500,000 protein structures for 99 fold-switching proteins in total. AlphaFold identified fold switching in 32 of 92 such proteins that are likely to be within its training set, meaning the model could have learned that they switch. The one successful identification of fold switching among seven proteins outside its probable training set had a related fold-switching protein among those used for training.
Porter and her colleagues say that this means that AlphaFold is unlikely to predict many new fold-switching proteins. They were also able to investigate the AI model’s different algorithms, finding they favoured information from pattern recognition processes over sequence coevolution.
Brenda Rubenstein from Brown University in the US, says that the study brings ‘much-needed’ rigour to understanding how AlphaFold and related algorithms work. ‘I applaud any efforts that attempt to better understand these algorithms so that we can advance not only these algorithms, but the field as a whole,’ she says. But she also stresses that they do currently work well, with her team successfully using them to predict challenging protein structures. ‘I wouldn’t bet too staunchly against machine learning models in general,’ Rubenstein says. ‘There is much room for improvement.’
Porter’s team also believes that AlphaFold can sometimes be useful in predicting fold switching, which could be an important advance. They’re also studying the biophysics and evolution of fold switching ‘so that better predictive models can be made in the future’, Porter says.
References
D Chakravarty et al, Nature, 2024, DOI: 10.1038/s41467-024-51801-z












No comments yet