Researchers behind a new AI-assisted technique that designs binding proteins to latch onto previously inaccessible drug targets are calling it a ‘major breakthrough’ in protein design. By targeting intrinsically disordered protein regions, which are common and often have roles in disease, including cancer and neurodegenerative conditions, the approach could pave the way for new treatments and diagnostic tools.
Around half of all proteins in the human proteome contain disordered regions that fold into variable, unpredictable structures. A bit like targets moving at random, these regions have long evaded scientists’ attempts to get drugs and antibodies to bind with them. Usually, proteins are targeted by designing small molecules that will always fit and bind to a given fixed ‘pocket’ shape in the folded protein’s structure. But in disordered regions, the shape of such pockets, if they are even present, are always changing.
Now, David Baker, who scooped half of last year’s Nobel prize in chemistry for his pioneering work on computational protein design, and his colleagues at the University of Washington, Seattle, US, have overcome this challenge with the help of AI.
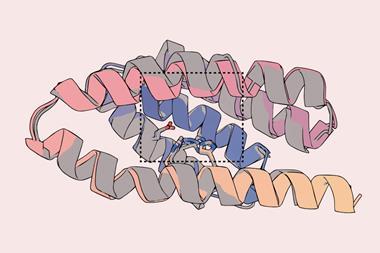
The team initially took inspiration from designed ankyrin repeat proteins (DARPins), which are a type of genetically engineered repeat protein scaffold that are derived from ankyrin, one of the most common families of binding proteins in nature. As such, DARPins usually exhibit highly specific and high-affinity protein binding.
‘DARPins have existed for decades, allowing people to screen for supposedly arbitrary folded protein targets with a huge phage library and get tight binders,’ explains co-lead author Kejia Wu, a postdoc in Baker’s lab. ‘I was curious whether a system like this could exist for fully disordered regions, but without the tedious library screening. Intuitively, I wouldn’t think it was possible due to the large conformational uncertainties. And if it was, how come nature hadn’t found it through evolution?’
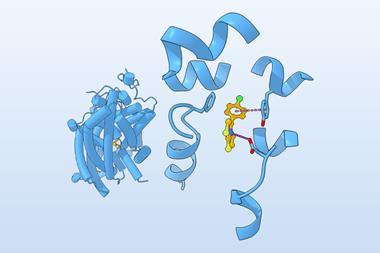
Despite what intuition said, the researchers set out to see what was possible. They came up with a design strategy called ‘logos’, which used generative AI to design and assemble binding proteins from a library of 1000 prefabricated parts, or binding pockets. The system was designed to mix and match these pre-made parts, allowing for trillions of combinations, to produce tailor-made binders that could potentially latch onto or wrap around almost any disordered protein or peptide target.
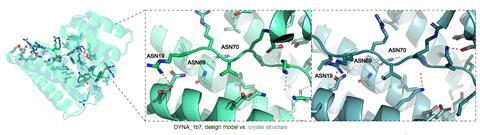
Of the 43 targets tested by the team, 39 of them produced tight binders. One aimed at the opioid peptide dynorphin was shown to block pain pathways in cultured human cells. Binders were also made for three signalling proteins linked to cancer. The technique was also shown to target and fluorescently tag a cell-surface protein linked to cancer, demonstrating how it could aid screening or cellular imaging of certain diseases.
Binding beauty
‘It was very surprising to us that it worked,’ says Wu. ‘I guess it says something about interesting biophysical properties in the protein sequence space that we didn’t, and probably still haven’t, fully understood, as well as the beauty in rational design.’
Baker adds that this is a ‘major breakthrough’ in protein design. ‘This is an important step as it is now possible to specifically recognise and drug a large, important class of proteins,’ he says.
‘[This work] builds on the very nice concept of binding modules in repeat proteins and explores these scaffolds for binding of diverse extended peptide conformations,’ comments Birte Höcker, who heads a protein design lab at the University of Bayreuth, Germany. ‘Many binders are shown to have high affinity for their targets. [But] how specific this is and whether very similar targets can be discriminated is not fully explored.’
‘I personally would have been interested to see more on specificity-determining factors and also how well binding pockets for peptide side chains could be achieved,’ says Höcker. ‘Nonetheless, it is an interesting step forward to binding intrinsically disordered regions.’
Pranam Chatterjee at the University of Pennsylvania, US, who published a method to design binders in January, says aspects of the latest work are ‘clever and well-engineered’. However, he remarks that the ‘targets the authors used are mostly short peptides (8–40 residues) with inherent structural propensity, not true disordered proteins’. Conversely, Chatterjee’s approach generates binders directly from a protein’s amino acid sequence using protein language models without requiring structural assumptions.
Baker acknowledges the logos method is limited by the length of the disordered region that can be targeted. However, he says they have developed another, complementary computational technique that overcomes this by also targeting amino acid sequences, rather than structural conformations, which is described in preprint and soon to be published in a prominent peer-reviewed journal.
References
K Wu et al, Science, 2025 DOI: 10.1126/science.adr8063









No comments yet