A team at Merck & Co has developed a five-step method for synthesising uprifosbuvir with a 50-fold increase in yield over the previous manufacturing process. The scalable strategy minimises undesirable side products and could be adapted to synthesise a range of nucleosides to help meet global demand for antiviral therapies.
Uprifosbuvir treats chronic hepatitis C infections. It is a nucleoside-based molecule that inhibits a specific RNA polymerase, thereby limiting viral replication. The original route to uprifosbuvir requires 12 individual steps, some suffering from poor regio- and diastereoselectivity, resulting in a 1% yield of the final product. By shortening the synthetic route to five steps and carefully optimising reaction conditions to maximise selectivity, the Merck team, led by Artis Klapars, has achieved a 50% overall yield when making over 100kg of the compound.
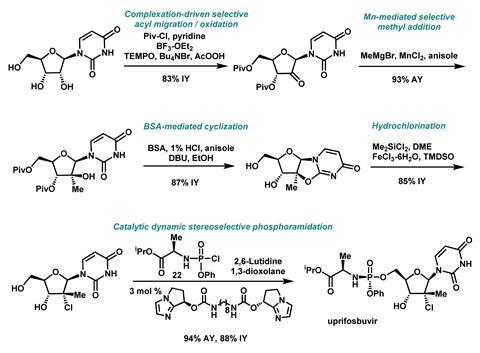
The team chose a readily available uridine as the starting material for the nucleoside component. A challenging functionalisation was then required in the 2′-position. They then explored a commercially-viable protecting group strategy using acyl chlorides, and by carefully selecting the solvent and Lewis acidic additives, the desired protected isomer was afforded before oxidation to the necessary ketone. This method of complexation-driven selective acyl migration and oxidation allowed 50:1 selectivity from the uridine starting material.
Methyl and chloro substituents were then introduced in the 2′-position via an olefination and hydrochlorination sequence. The process of installing the methyl substituent was then adapted to improve cost, safety and environmental impacts, by using an organomanganese reagent. To selectively convert the tertiary alcohol, the team hit upon FeCl3·6H2 O and tetramethyldisiloxane as inexpensive reagents to drive the equilibrium towards the tertiary alkyl chloride with high diastereoselectivity, unveiling the full nucleoside structure of uprifosbuvir.
Finally, a phosphoramidate side chain was attached using a specially designed chiral nucleophilic imidazole carbamate catalyst, delivering uprifosbuvir in an impressive 50% overall yield.
‘The most challenging step in terms of optimisation was the complexation-driven selective acyl migration and oxidation because of the number of transformations that are interrelated there,’ highlights Klapars. ‘Reaction selectivity is certainly an important factor. Ensuring robustness and scalability of reactions is one of the guiding principles of our work. We go through great lengths to understand how the reactions work and to define operating ranges that deliver quality product on any scale required.’
Katherine Seley-Radtke, an expert in the design of nucleoside drugs at the University of Maryland, Baltimore County in the US, is impressed by the improved protocol. ‘They have managed to take something that was rather complex and make it very simple, very straightforward and very high yielding with readily accessible materials. They went from extremely poor yields to a 50-fold increase in yield. I think the major aspect is the stereospecificity that they were able to achieve. I think it might have tremendous impact.’
Erik De Clercq from the Rega Institute for Medical Research in Belgium, a co-inventor of some of the first anti-HIV medications, also says the method could have broad applicability. ‘The reported approach may show utility in the derivatisation of antiviral agents at large and certainly C-nucleoside analogues such as remdisivir and others such as pyrazofurin.’










No comments yet