
Selective cross coupling of carbonyls to form alkenes, especially non-symmetrically, is an important reaction in organic synthesis. However, only a few methods are available and none provide efficient cross-selectivity in carbonyl–alkene reactions. Now, an international team has introduced two strategies for cross-metathesis of carbonyls, enabling carbene-to-ylide transfer and di-metalation to make alkenes.
David Nagib at Ohio State University in the US, who developed the methods with Lumin Zhang from the Shanghai Institute of Organic Chemistry in China, traces the origins of this idea back to 2005, when the chemistry Nobel prize was awarded for the development of metathesis in organic synthesis. ‘I was just a young, naive student,’ Nagib recalls. ‘I thought, “What if you could take two carbonyls and mix them together and make it an alkene in the same process?” And obviously, there are many difficulties for doing it. But it’s been in the back of my mind since 2005.’
The first strategy involved converting aldehydes into phosphonium ylides through a modified Wittig mechanism. This approach led to the formation of Z-alkenes with high stereoselectivity. Nagib and Zhang demonstrated the effectiveness of this method by successfully converting enolisable alkyl aldehydes into Z-alkenes with cross-selectivity and stereoselectivity.
The second strategy drew inspiration from the Takai reaction, which involves the stereoselective olefination of aldehydes using di-metals. In this approach, the duo used chromium catalysts and manganese reductants to generate dichromium intermediates, driving alkene formation and carbonyl deoxygenation. This method resulted in the formation of E-alkenes with high selectivity.
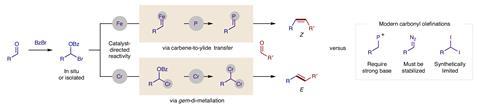
An important property of both mechanisms is their efficient and economical cross-selectivity. In traditional cross-reactions, chemists often use an excess of one substrate to drive the reaction towards the desired outcome. But that’s not the case here. ‘In our strategy, we only use 1:1 equivalent [of substrates] to achieve cross-selectivity,’ adds Zhang.
When comparing the new strategies to other state-of-the-art methods, Vincent Lindsay from North Carolina State University sees a clear frontrunner. He tells Chemistry World the McMurry coupling strategy developed in the 1970s is only efficient for either dimerisation processes forming symmetrical alkenes or for intramolecular couplings to achieve cyclic derivatives. He says that Nagib and Zhang’s strategies are a step forward.
‘Considering that most desirable alkenes in organic synthesis are not symmetrical, this work is absolutely transformative and significantly expands the retrosynthetic possibilities available to chemists for the elaboration of alkenes,’ he says.
He also adds that the fact that both Z- and E-isomers can be independently prepared in a catalytic manifold from the same substrates is ‘incredibly attractive, enabling this novel disconnection to efficiently compete with classical transforms for alkene synthesis such as the Wittig, Horner–Wadsworth–Emmons or Julia olefinations, among others’.
References
L Zhang and D Nagib, Nat. Chem., 2023, DOI: 10.1038/s41557-023-01333-8












No comments yet