What determines how reaction rates respond to thermodynamic driving forces? And why do some reactions speed up dramatically with slight changes in driving force, while some others appear much less responsive? Hoping to answer questions like this, theoretical chemists in Germany have introduced a new framework to model the reactivity landscapes of chemical reactions.
Understanding the role of external factors in chemical reactions is central to theoretical and experimental chemistry research. Gaining deeper insight into these factors can help chemists steer reactions toward desired products, maximise yields and limit unwanted side reactions.
Chemical reactions can be viewed as a continuous search for stability. From a thermodynamic perspective, this stability is measured by Gibbs free energy, and a reaction typically progresses along a path that lowers this energy. However, this path isn’t unique – reactions can evolve through multiple suboptimal configurations depending on external influences.
Classical models, such as the Leffler or Marcus equations, have attempted to capture how reaction rates respond to external driving forces, aiming to identify favoured reaction routes. However, these models are constrained by the spectrum of thermodynamic situations they can describe. More advanced statistical approaches, while broader in scope, often rely on multiple reaction-specific parameters, which can reduce their general interpretability.
A new equation with a single parameter
Now, Eduardo García-Padilla and Guanqi Qiu at the Max Planck Institute for Coal Research in Germany have proposed a model that links a reaction’s activation energy (ΔE‡) to its overall reaction energy (ΔEr). Their equation relies on a single parameter – the curvature factor θ – which reflects the stability of the transition state and how it’s influenced by competing factors affecting the reaction.
Unlike previous approaches, the model follows key properties of chemical reactions without resorting to restrictive assumptions. The result is a framework that maintains the accuracy of statistics-based models while retaining the simplicity of classical models.
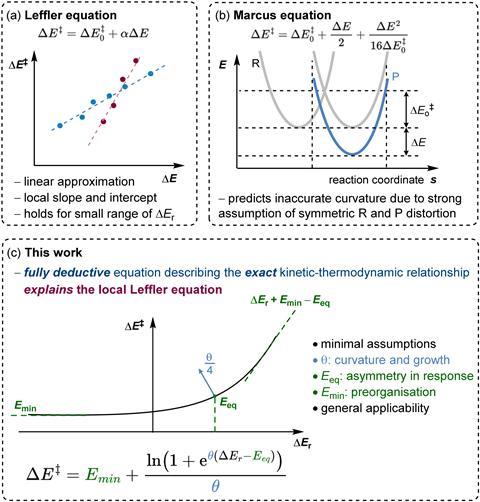
García-Padilla predicts that their model will help chemists uncover new reaction pathways by revealing hidden relationships between the forces driving chemical transformations. ‘Some of the parameters tell you, for example, that there is a minimum activation energy that will never be able to be overcome just by making the reaction more favourable. This already gives synthetic chemists more control over the reaction than assuming there is a linear free energy relationship, and that the reaction will always respond in the same way.’
García-Padilla envisions a future where predicting model parameters for a family of reactions will allow users to predict ‘how much a reaction will respond and tune the response to increase the selectivity between two possible processes’ under an applied driving force.
Looking ahead, García-Padilla thinks machine learning will emerge as a natural alternative to predict the model parameters within a certain family of reactions. ‘These sorts of combinations of machine learning to predict not the reaction rate but some physically useful parameters would be very interesting. Even if the machine learning parts would be sort of black box, the good thing is that the model parameters themselves, you can easily check their validity.’
‘The [study] constitutes a timely and impactful contribution to the fields of computational chemistry and reaction design,’ says Marco Martinez, at expert on theoretical chemistry methods for modelling chemical reactivity at McMaster University in Ontario, Canada. ‘Determining reaction barriers requires the optimisation of the reactant and transition state connecting to the product. This makes the process time-consuming and highly dependent on human expertise, hence, unsuitable for high throughput screening approaches. In contrast, the proposed model can be parameterised with existing data from related reaction families. This makes it suitable for high throughput screening, offering the best of both worlds: low computational cost and physically meaningful predictions, allowing to better identify the driving force behind reaction kinetics.’
Martinez says the model requires additional analysis and validation. ‘The model parameters are related to chemically interpretable concepts with no definitive relationship between them and chemical properties of the reactants and products,’ he explains. ‘Additionally, the model’s performance should be assessed for highly asynchronous reactions or in cases with multiple competing reaction paths with similar barriers.’
References
This article is open access
E García-Padilla and G Qiu, Chem. Sci., 2025, 16, 17494 (DOI: 10.1039/d5sc04829j)









No comments yet