BRSM wonders what makes a route so good it becomes the last total synthesis of a complex target
Chemists do total synthesis for many different reasons, and therefore there are many criteria by which a route can be judged successful. Getting to the target is important, but if you’re the fourth person to make a popular natural product, you’ll need to do something really interesting to stand out. There are many metrics to assess syntheses: atom economy, ideality, step economy and – the process chemists’ favourite – product mass intensity, to name just a few. However, you’ll rarely hear these mentioned in an academic setting. Creativity – and the elusive ‘elegance’ – are usually considered higher goals, although harder to assign numerical values.
It’s sometimes said that for a synthesis to be remembered, it needs to be either the first or the last. Popular natural products often result in furious races between research groups to finish them first – that’s easy to understand. The aim of being last is to achieve such a spectacular synthesis that no one else will be able to do any better, and hence no one else will even try.
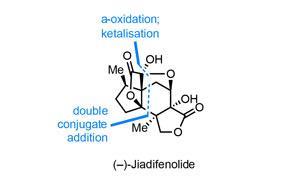
Perhaps unsurprisingly, first syntheses are vastly more common than last, and of course the latter is much more subjective. That said, I believe that Ryan Shenvi’s could well be the last synthesis we see of the popular neurotrophic agent jiadifenolide, at least for some time.1 Using a daring double conjugate addition reaction to unite two much more manageable fragments, Shenvi and co-workers at the Scripps Research Institute in La Jolla, US, complete the complex target in a total of just 10 steps, far surpassing previous routes in brevity, efficiency and elegance.2
The team begins with the fragrant and inexpensive (+)-citronellal, converting it into a bicyclic butenolide in three steps (figure 1). Preparing the second building block (a ß-ketoester) from hydroxyacetone and trimethyldioxinone is even shorter – requiring just two steps – setting the stage for the crucial double conjugate addition reaction.
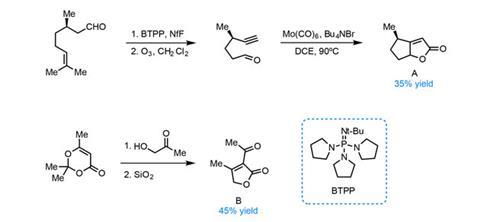
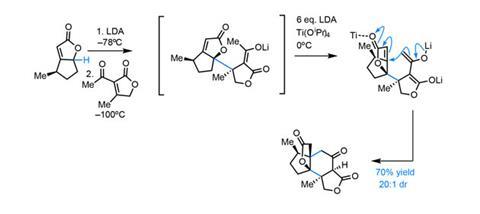
The first conjugation reaction proceeds with excellent selectivity, guided by the single citronellal-derived stereocentre (figure 2). Low temperatures maintain selectivity in both the deprotonation and addition. Adding a further portion of lithium diisopropylamide (LDA) deprotonates the resulting enolate a second time, allowing the second conjugate addition to close the central six-membered ring.
This unusual coupling reaction provides rapid access to the complete carbon skeleton in good yield. Incredibly, the single stereocentre in the starting materials unerringly directs formation of four more, giving the stereochemically-dense product as essentially a single diastereomer.
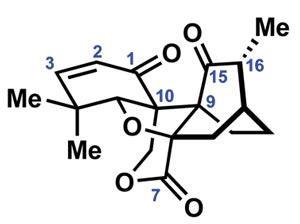
The group needs just four redox tweaks to complete the natural product (figure 3). First, hydroxylation of the carbon between the ß-ketoester carbonyls proceeds smoothly using m-chloroperbenzoic acid. The new hydroxyl group then directs reduction of the adjacent ketone using [Me4N]BH(OAc)3, selectively giving the trans-1,2-diol. Finally, a two-step protocol involving bromination of the enolate trianion, followed by oxidation with Davis oxaziridine triggers the final ring closure.
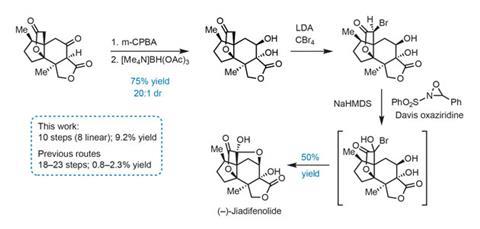
Using this route, the group has now prepared grams of this scarce molecule. They are making it available to researchers interested in studying its neurotrophic properties, keeping a promise of solving supply issues that is so often broken by synthetic chemists.
BRSM (@BRSM_blog) is a postdoctoral researcher and blogger based in the US
References
1 H-H Lu, M D Martinez and R A Shenvi, Nat. Chem., 2015, 7, 604 (DOI: 10.1038/nchem.2283)











No comments yet