James Mitchell Crow wonders what would make the perfect organic synthesis

In the ongoing hunt for better cancer treatments, the bottom of a boat might not sound like a promising place to start. But that is where you might find a seaweed-like organism called Bugula neritina happily growing. To the shipping industry, the organism is a troublesome foulant of ships, piers, and buoys exposed to warm marine waters. But Bugula neritina also produces some of the most potent anti-cancer compounds known.
Just 1.2 milligrams of bryostatin 1, as the most prized member of the bryostatin family is known, taken over eight weeks is all that is needed to potently enhance the effect of chemotherapy, clinical trials have shown. The compound also shows promising activity in enhancing cognition and memory in animal models of Alzheimer’s disease, but efforts to develop byrostatin-based drugs are hampered by the molecule’s restricted natural supply. To carry out initial clinical studies on the compound, 14 tonnes of Bugula neritina had to be processed in order to generate just 18 grams of material.1
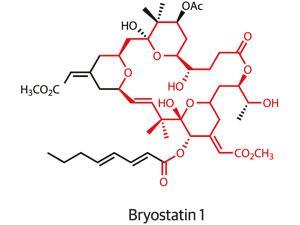
It is cases like these where the natural product total synthesis community would hope to step in, offering an alternative source of a molecule by building it, step by step, from simple and readily available starting materials.
Unfortunately, as well as their promising bioactivity, the bryostatins are also renowned for their structural complexity. Their macrocyclic structures are densely packed with side-chains and functional groups. To date, the long sequence of chemical transformations needed to synthesise bryostatin 1 — 57 steps — has meant that total synthesis has failed to provide useful quantities of the natural product.
Such examples are one of the main drivers behind the increasingly popular philosophy of ‘ideal synthesis’. Given enough time and manpower, the field of organic synthesis has matured to the point where even the most complex target molecules can be made. The focus of organic chemistry today is increasingly switching toward making them in a practical, scalable manner.
Ideas of the ideal
Achieving efficiency in synthesis is clearly a challenge in itself – but only one of the parameters that an ideal synthesis should achieve. Green chemistry principles are also encompassed, for example. Paul Wender at Stanford University in the US codifed many of the concepts collectively thought of as ideal synthesis. In a book first published in 1993,2 Wender proposed the following definition: ‘The ideal synthesis may be defined as one in which the target molecule is prepared from readily available starting materials in one simple, safe, environmentally acceptable and resource-effective operation that proceeds quickly and in quantitative yield.’
Wender acknowledges that the definition is an aspirational one, but speaks passionately about its value for pushing synthesis forward. ‘By benchmarking ourselves to what others before us have done, we’re always going to come out looking good because we have more tools and more knowledge — and that’s how one can get caught in incremental change,’ he says. ‘I think we can leap forward faster by thinking about what’s possible and what’s ideal.’
There is more than one definition of what an ideal synthesis might look like, though. Phil Baran at the Scripps Research Institute in La Jolla, US, places a strong emphasis on ideality in his own work in natural product synthesis. ‘There are gradations of ideality,’ he says. ‘There’s “Star Trek” ideality where you start from charcoal and make [highly complex natural product] brevetoxin in one step.’ At the other end of the scale is what Baran refers to as ‘real world ideality’, which he says is ‘something that you can teach a student, for them to develop a synthesis that is much more efficient than if they hadn’t considered the idea’.
Interpretations may vary, but ultimately the goal is the same, he adds. ‘We all agree that if you can make a complex molecule in an academic setting in a scalable fashion, with a minimum amount of resources, then that must be a pretty good synthesis.’
The essence of ‘real world’ ideal synthesis is captured by the definition proposed by James Hendrickson from Brandeis University, US, in 1975, Baran says.3 Hendrickson defined the ideal synthesis as one which ‘creates a complex molecule…in a sequence of only construction reactions involving no intermediary refunctionalisations, and leading directly to the target, not only its skeleton but also its correctly placed functionality.’4
‘Step economy’ is one aspect of ideal synthesis, but ideality demands other attributes as well, says Barry Trost, also at Stanford. Trost emphasises ‘atom economy’ — the idea of designing a transformation to carry as much of the reactants’ mass as possible into the product, maximising sustainability by minimising waste. ‘On some fronts there has been very significant progress [toward ideal synthesis], but the area where I don’t think we have come a long way is people latching onto atom economy as being as important as it should be,’ says Trost.
As Hendrickson’s near-40-year-old publication shows, the concept of ideality in organic synthesis is not new. ‘A lot of this has been part of the culture of synthesis from the get-go,’ says Wender. ‘If you look at what most people in the field get excited about, it is solutions to problems that are very concise.’ What has refocused attention on it in the last few years has been the emerging possibility of making significant steps towards ideality, given the rapidly expanding arsenal of reactions that organic chemists can now bring to bear on a complex natural product. Whereas today the challenge is to synthesise members of the bryostatin family more efficiently, not so long ago the challenge was simply to synthesise them at all.
Practical ideal synthesis
Philosophy aside, what is the best approach to design an efficient synthesis today? To maximise step economy, reactions that fail to meet Hendrickson’s definition of ‘construction reactions’ – steps that directly contribute to the building of the target molecule’s skeleton — are the ones to avoid. These reactions come in a few forms, says Baran, who refers to them collectively as ‘concession steps’.

The first concession step category is the non-strategic redox manipulation – for example, reducing an ester group down to an alcohol, only to oxidise it back up to an aldehyde later in the synthesis. An extra step or two here and there might not sound like a big problem, but the impact is cumulative. Every synthetic step inevitably involves a loss of material, because very few synthetic transformations give solely the desired product. Purification processes at the end of a reaction invariably introduce losses too. To illustrate the scale of the problem, a kilogram of starting material subjected to a 20-step synthesis, each step giving an 80% yield, would result in only 11 grams of final product. And the majority of natural product syntheses will have at least a couple of steps yielding significantly less than 80%, and many syntheses need more than 20 steps.
The next kind of step to avoid is protecting group manipulation. Protecting groups are used to mask reactive functional groups, allowing them to be carried through the synthesis without reacting in undesirable ways. However, adding the protecting group and then removing it later adds extra steps.
Baran has been a leading proponent of protecting-group-free synthesis. ‘They are a necessary evil sometimes, but I think that ever since we and others started putting it forward that you could consciously exclude protecting groups from the way that synthetic designs are formulated and executed, there’s been a large number of synthesis and methodologies saying, let’s see what happens when we leave the protecting groups out.’ In many cases, protecting groups have successfully been avoided.
The third and final group of undesirable steps in a synthesis are functional group interconversions. Functional groups often serve as the handles by which the molecule’s carbon skeleton is clipped together. However, if the functional groups used in this way are not the ones found in the target molecule, then a potentially costly series of interconversion can be needed late in the synthesis, switching an alkene for a ketone, say.
Unfortunately, avoiding all such steps entirely is rarely achievable, Baran acknowledges. ‘We apply the term concession step to these types of reactions since it is well accepted that they require extra effort but are often simply unavoidable.’
However, they might not always be so unavoidable. New synthetic methodologies continue to appear that offer ways to trim away these unproductive steps, and perhaps the most promising is selective C–H bond activation.
‘If you’re going to try to aim for an ideal synthesis, one of the main things is you want to avoid functional group manipulations — and the easiest way to avoid those is to avoid functional groups,’ says Baran. ‘C–H functionalisation is one main vehicle to achieve that.’
The average organic molecule bristles with C–H bonds — so much so that when an organic structure is drawn out, the hydrogen atoms are ignored for clarity. The ideal way to attach a new functional group to a molecule would be to simply pluck off the relevant hydrogen atom and add the new group in its place. The sheer number of C–H bonds in a molecule has historically made this process all but impossible. However, a series of discoveries in the past five years have started to show that it can sometimes be done, with the right catalyst — and the approach is already beginning to prove its worth in the total synthesis arena.5 ‘It’s a nascent field,’ says Baran. ‘I think there are a tremendous number of things waiting to be discovered in that area.’
Wender agrees. ‘I think C–H activation is hugely important as it allows us to reinvent, if you will, functional groups. And not just C–H activation, but C–C activation and C–heteroatom activation: all of these things will be of consequence.’
New chemistry needed
Trimming concession steps from a synthesis is important, Wender says – but to cut more than a few steps from a synthesis, other approaches are needed. ‘If you really are striving to come up with the best synthesis, that often requires the invention of new science,’ says Wender. ‘It’s the difference between incremental and transformative change.’
Baran agrees that new chemistry is the key to ideal synthesis, as well as one of its main attractions. ‘Ideal syntheses are often a little bit more difficult to plan, because they often require invention, but I think that’s the whole point,’ he says. ‘If you do synthesis just with the idea of applying known things, it’s not awfully exciting.’
As a recent example of the way that a newly discovered reaction can revolutionise synthetic strategy, Wender points to the olefin metathesis reaction, a powerful way to clip molecular fragments together by forming a new carbon–carbon double bond, which earned its inventors the 2005 Nobel prize in chemistry.
Wender’s lab has pioneered another approach for quickly turning simple starting materials into much more complex molecules. Known as the arene–alkene meta-photocycloaddition, it introduces three new bonds and up to six chiral carbon centres in a single step. ‘Almost across the board, this reaction has led to the shortest syntheses of the molecules that we have looked at,’ says Wender. The original synthesis of the natural product cedrene, for example, took 25 steps. ‘Our synthesis took four steps,’ he says.6
‘There’s no surprise in this — it has nothing to do with our creativity,’ Wender adds. ‘If you have new reactions that allow for a great increase in complexity, they’re going to lead to shorter syntheses no matter who uses them.’
There is no doubt that more such reactions are still waiting to be discovered. ‘The teacher that I often cite is nature,’ says Wender. ‘What we’re seeing there is very enlightening — compartmentalisation is very important, cascade reactions are very important. These are things that we will eventually master and they will have a transformative impact.’
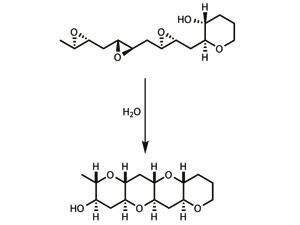
Cascade reactions are already beginning to appear in total synthesis, and have proven particularly useful for synthesising a group of marine natural products known as ladder polyethers,7 which are thought to be made this way by nature. In one key step, the entire molecule is zipped together as bond after bond snaps into place, the product of one bond-forming process immediately reacting to form the next.
Non-traditional approaches to synthesis also offer significant promise, Wender adds. Flow chemistry offers one synthetic equivalent to nature’s compartmentalisation approach, where a molecule is passed from reaction chamber to reaction chamber within a flow synthesis device without ever needing to be isolated, until the final product comes out at the end.
Biocatalysis is another field with considerable potential, as biochemists become increasingly adept at reengineering enzymes to catalyse particular transformations, says Wender. Nick Turner, a biochemist who leads the University of Manchester’s Centre of Excellence for Biocatalysis, Biotransformations and Biocatalytic Manufacture, points to the recent emergence of enzymes called transaminases, which convert ketones directly into chiral amines. ‘There really isn’t equivalent chemistry to do that transformation in a single step,’ says Turner. Without enzymes, this process requires four separate steps to complete. US pharmaceutical company Merck and Co, alongside collaborators at biocatalysis company Codexis, recently developed a transaminase for the final synthetic step of its type 2 diabetes drug Januvia (sitagliptin phosphate). Given that around 40–50% of pharmaceutical compounds currently in development incorporate a chiral amine group, it’s the kind of chemistry that will have broad impact, Turner says.
Such developments aside, total synthesis is still some way from ideality, and molecules such as bryostatin stand as a formidable challenge. However, the journey is already generating improved ways to make molecules. ‘We may never achieve a total synthesis characterised by 100% ideality,’ says Baran, ‘but such a pursuit serves as a constant source of inspiration.’
James Mitchell Crow is a science writer based in Melbourne, Australia
References
1 D E Schaufelberger et al, J. Nat. Prod., 1991, 54, 126 (DOI: 10.1021/np50077a004)
2 P A Wender and B L Miller in Connectivity analysis and multibond-forming processes in Organic Synthesis: Theory and Applications (T Hudlicky, ed.), 27–66. JAI Press, 1993
3 T Newhouse, P S Baran and R W Hoffmann, Chem. Soc. Rev., 2009, 38, 3010 (DOI: 10.1039/b821200g)
4 J B Hendrickson, J. Am. Chem. Soc., 1975, 97, 5784 (DOI: 10.1021/ja00853a023)
5 E M Stang and M C White, Nat. Chem., 2009, 1, 547 (DOI: 10.1038/nchem.351)
6 D Chappell and A T Russell, Org. Biomol. Chem., 2006, 4, 4409 (DOI: 10.1039/b614011b)
7 I Vilotijevic and T F Jamison, Science, 2007, 317, 1189 (DOI: 10.1126/science.1146421)











No comments yet