The potential of these targets has not gone unnoticed in the synthetic community, nor has the stereochemical complexity of the tetracyclic system. However, only one total synthesis had been achieved3 when Seth Herzon and his team at Yale University, US, set out on their synthetic odyssey, leaving plenty of unexplored options to investigate.4
The team start with a fragment reported in a prior publication,5 developed using an extremely neat strategy combining Diels–Alder and Staudinger reactions.
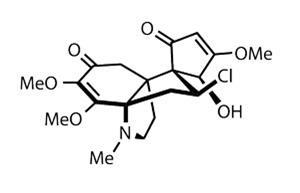
The team employs the former of these two statesmen of the named-reaction canon, the Diels–Alder cycloaddition, to combine a quinone with a silyl cyclopentadiene, using a chiral mediator to induce asymmetry in the product (figure?1).
Better still, adding a little trimethylphosphine kicks off that intramolecular Staudinger reaction, named after the father of polymers, German chemist Hermann Staudinger. This reaction is particular to azides, transforming these energetic tri-nitrogen species into imines by reaction with carbonyl groups.
In this case, the Staudinger builds complexity by adding a further heterocycle to create the fragment with which the latest paper begins.
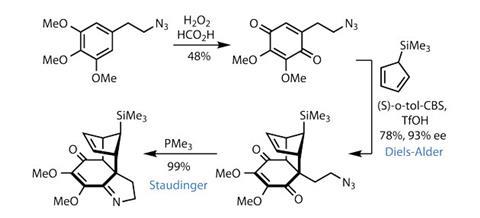
Working directly with the freshly-introduced imine functionality, the group methylates the nitrogen and bolts on a large cyclopentene-based sidechain derived from D-ribose, a common sugar. An acetylene functionality in the sugar-derived intermediate allows the team to bring in this sidechain by simply deprotonating the terminal acetylene and adding it to the imine. The team then removes the silyated cyclopentene by retro-cycloaddition, delivering a cyclohexadiene moiety ready for further reaction.
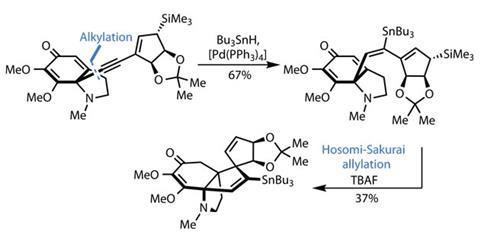
Hydrostannylation with tributyltin hydride and a simple palladium catalyst transforms the acetylene group into a vinylstannane, providing a flexible framework for a Hosomi–Sakurai allylation (figure?2). This less well-known reaction employs allyl silanes as activated partners to add into enone groups. While the yield in this particular case is relatively low, the ease with which the team builds the tetracyclic system is remarkable.
Paul Docherty is a science writer based in Reading, UK











No comments yet