Palau'amine
Palau’amine
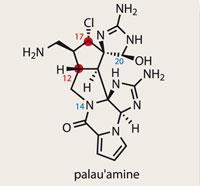
It’s not very often that a molecule is classed as ’un-makable’; it’s even rarer that it then finds its way into the total synthesis literature. Such an exotic beast is palau’amine, an alkaloid which has stubbornly held off synthesis for over 15 years. Its nemesis comes in the form of Phil Baran at the Scripps Research Institute, La Jolla, US.1
Numerous routes towards palau’amine have been documented in the literature, including impressive efforts by Larry Overman from the University of California, Irvine, US, and Erick Carriera at the Swiss Federal Institute of Technology (ETH) in Zurich.2 The crux of the problem is the cyclopentane at the heart of the molecule, which crams in a full complement of five stereocentres.
However, after 12 years of endeavour, a close examination of the structure of palau’amine revealed a stereochemical quandary. In a 2007 paper,3 the combined teams of Baran and Matthias K?ch from the Alfred Wegener Institute in Bremerhaven, Germany, reassigned a pair of stereocentres (C17 and C12). This brought the cyclopentane core into familiar territory, as it was similar to that of the axinellamines and massadines.
At this point, the Baran-led team must have been quietly confident, having published syntheses of members of both these families in 2008.4,5 But the reassignment brought both joy and fear, as the newly configured C12 centre suggested a trans-5,5-fused ring system, known to be particularly problematic.
Waste not, want not
Undaunted, the team carried on, recycling some of the chemistry from their synthesis of the massadine. A Diels-Alder reaction was used to begin with - a classic method of using ring formation to impart stereocontrol. Fragmentation of this bicyclic system left a cyclopentane with three of the required stereocentres. When this was treated with a hypervalent iodine oxidant (IBX), a neat cyclisation formed a 2-aminoimidazole ring and set a fourth, particularly congested stereocentre (figure 1). Completion of the cyclopentane fragment required only a few more steps, and it was here that the routes to palau’amine and massadine diverged.
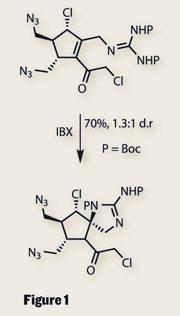
A second 2-aminoimidazole was easily installed, with bromination providing a key synthetic handle for forming a pyrrole ring. But before they did this, the team performed a remarkably selective oxidation using an unusual silver(ii)-picolinate reagent. This provided the C20 hydroxyl group, manifesting as part of an N,O -acetal. In keeping with Baran’s distaste for protecting groups, this was left unprotected in the remaining steps.
Tripping at the line
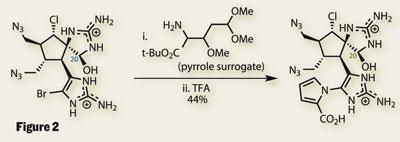
Having performed such an intricate reaction so effectively, the pyrrole synthesis was expected to be trivial. However, it turned out that a complex pyrrole surrogate was required (figure 2). Under acidic conditions, this reactive molecule attacks the bromo-imidazole and wraps around to form the pyrrole by a series of elimination reactions.
Completing the synthesis required only three more steps, all done in one pot. Reduction of the two precarious-looking azide groups revealed a pair of amines. The nearer of these was then coupled with the pyrrole carboxylic acid to provide a macrolactam - dubbed ’macro-palau’amine’ by Baran. Under acidic conditions, the amide nitrogen reaches across the macrocycle, forming the desired 5,5-fused system (figure 3). Perhaps the biggest surprise in this reaction is that a sniff of acid was all that was needed to make such a notoriously difficult system.
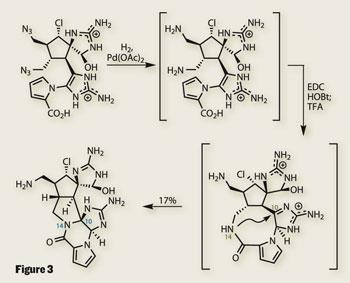
This elegant cyclisation was the coup de gr?ce in this superlative synthesis. With both palau’amine and vingrol6 completed within months of each other, Baran’s team must be looking forward to the total synthesis challenges presented by a new decade.
Paul Docherty is a medicinal chemist based in London, UK
References
1 P S Baran et al, Angew. Chem. Int. Ed., 2009, DOI: 10.1002/anie.200907112
2 S M Weinreb, Nat. Prod. Rep., 2007, 24, 931
3 M Köck et al, Angew. Chem. Int. Ed., 2007, 46, 6586
4 P S Baran et al, J. Am. Chem. Soc., 2008, 130, 16490
5 P S Baran et al, Angew. Chem. Int. Ed., 2008, 47, 3581
6 P S Baran et al, J. Am. Chem Soc., 2009, 131, 17066










No comments yet