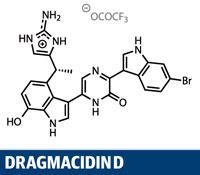
Why do we make functional groups? That might come across as the dumbest question of the year, but there’s actually a point behind it. The majority of functional groups present in the synthetic intermediates in a synthesis don’t make it to the target - they are consumed, mostly in coupling reactions to form carbon-carbon bonds. Furthermore, those key bond-forming reactions tend to be particular to the union of specific functional groups; carbonyl groups to perform aldol reactions, vinyl iodides and boronic acids for Suzuki couplings, and so on. So why not cut out the middleman, and couple the simple fragments? Well, C-H bonds tend to be very unreactive, and even the simplest alkyl or aryl fragments present a significant selectivity issue - how do you choose just one C-H out of the multitude? But these issues don’t deter chemists like the team led jointly by Junichiro Yamaguchi and Kenichiro Itami at Nagoya University in Japan.1
What we’re talking about here is C-H activation, an area of intense synthetic study and a buzzword bingo winner when applying for research grants.2,3 What the Japanese team demonstrates in this synthesis of dragmacidin D is the state of the art, uniting all the key fragments using C-H bond couplings. The target isn’t particularly new - a rather neat synthesis by Brian Stoltz and his team at the California Institute of Technology, US, was published in 2002, completing the target in a relatively efficient 17 steps.4 But by cutting out functional group transformations, the Japanese team has trimmed that back to an extremely economical 12.
The group started the campaign with one of the most venerable C-H activations - and by cheating slightly. The union in question was that of an iodoindole with a C-3-substituted thiophene (both derived from commercial starting materials in two or three steps), using a modification of the Heck coupling. The ’cheat’ was using a halide atom on the indole to direct the reaction. However, the group still had to overcome a selectivity issue with the thiophene, as both the C-2 and C-4 C-H bonds are reactive.
Flick the switch
The team overcame this issue by judicious choice of protecting group on the hydroxyl group. Using a small methyl group led to unselective reaction, but switching to the bulky triisopropylsilyl (TIPS) group blocked reaction at C-2 and gave the team a reasonable yield of the desired regioisomer (figure 1).
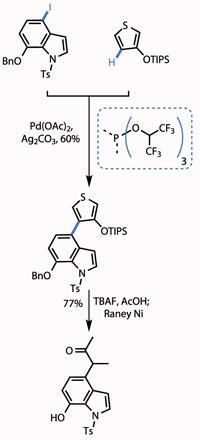
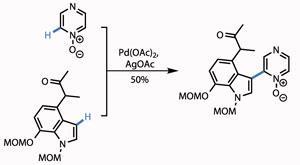
Treating the protected hydroxythiophene with tetrabutylammonium fluoride (TBAF) removed the bulky silicon-based protecting group, while reaction with Raney nickel (fine grains of an activated Al-Ni alloy) reduced the thiophene, unmasking a methyl ketone. A little protecting group shenanigans took them to the point of their next reaction - bolting on a pyrazinone ring. The team used the fact that pyrazinones are in equilibrium with pyrazine N -oxides, facilitating a palladium catalysed coupling of two unfunctionalised C-H bonds in a satisfying yield (figure 2).
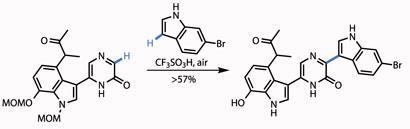
Simply treating the resulting N -oxide with trifluoroacetic anhydride switched the isomers, revealing the pyrazinone group, readying the team for the final coupling. This was to append a further indole moiety, using an oxidative Friedel-Crafts-type reaction, mediated by trifluoroacetic acid. Happily, this also removed both methoxymethyl (MOM) protecting groups from the other indole section (figure 3).
Two further reactions were required to get to the target: brominating the methyl ketone to provide a synthetic handle (a rare example of functional group transformation in this synthesis), then displacing that bromine with Boc-protected guanidine, leading to cyclisation with the ketone.
Although not entirely devoid of functional group transformations, this synthesis and those like it point towards an ideal strategy, where only coupling reactions are required. However, C-H activation is still in relative infancy - we’ve come a long way, but there’s so much more to do.
Paul Docherty is a science writer and blogger based in Reading, UK
References
1 D Mandal et al, J. Am. Chem. Soc., 2011, DOI: 10.1021/ja209945x
2 D Alberico, M E Scott and M Lautens, Chem. Rev., 2007, 107, 174 (DOI: 10.1021/cr0509760)
3 H M L Davies and J R Manning, Nature, 2008, 451, 417 (DOI: 10.1038/nature06485)
4 N K Garg, R Sarpong and B M Stoltz, J. Am. Chem. Soc., 2002, 124, 13179 (DOI: 10.1021/ja027822b)










No comments yet