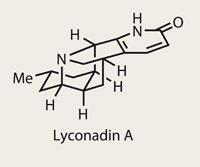
Having spent a few years working in the pharmaceutical industry, one thing I’ve learnt is that Alzheimer’s disease isn’t an easy nut to crack. Any opening into its prevention or treatment is understandably leapt upon, especially natural products such as the Lycopodium family that show activity against the disease. Members such as huperzine A are already in use as treatments in China, with the rest of the family under intense scrutiny. However, some, such as lyconadin A, are naturally available in only vanishing quantities, so a synthetic source is enormously significant - as well as a guaranteed funding proposal winner. Richmond Sarpong’s research group at University of California, Berkeley, US, have taken quite an interest in lyconadin A, publishing an initial, racemic synthesis in the form of a communication;1 now they have added to that result with an enantioselective route, using some of the same ideas, but refining the process along the way.2
As is often the case with tightly functionalised targets containing several adjacent stereocentres, the group hoped that setting one stereocentre would allow ‘substrate controlled’ definition of the others. However, while high yielding, initial attempts using this approach gave the wrong diastereomer of product. The team used an interesting strategy to get around this problem - creating a temporary stereocentre with ease, and using that to control further asymmetric chemistry (figure 1). The stereocentre in question was installed via a CBS (Corey-Bakshi-Shibata) reduction, creating a chiral secondary hydroxyl in high yield and enantiomeric excess. This alcohol was then able to direct a double hydrogenation, reducing two alkenes to generate the three required stereocentres in excellent diastereomeric excess. Its task dutifully completed, the hydroxyl group was then oxidised back to its initial ketone state.
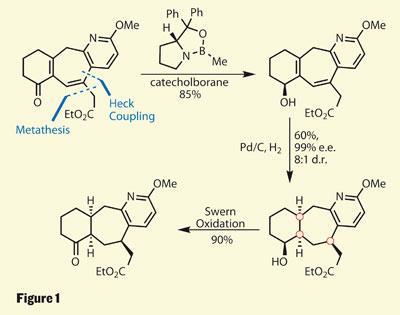
Reductionism
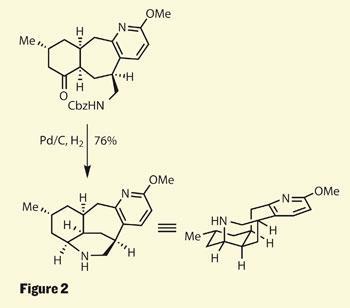
The next key transformation was yet another reduction, this time to remove a labile carboxybenzyl (Cbz) protecting group from a primary amine. This reaction doesn’t actually cause a cyclisation in its own right, but allows the free amine to attack the ketone, forming an intermediate imine which was itself reduced, creating one more stereocentre (figure 2). Again, substrate control provided the new stereocentre as a single diastereomer.
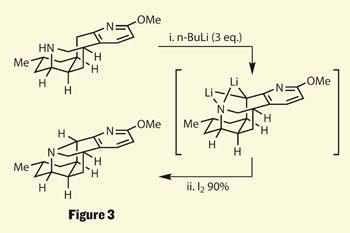
As this point, the group were tantalisingly close to the target - only one carbon-nitrogen bond away. However, there appears to be a distinct lack of functionality with which to build this bond, and my first impression was one of confoundment. But after reading about the chemistry used, I feel less chemically inadequate - anyone heard of the Boekelheide variant of the Polonovski rearrangement?3 Or the Hofmann-Löffler-Freytag reaction?4 I hadn’t - until now - so even though these approaches failed, their discussion is one of the things that makes this such a great paper. But it wouldn’t be here unless they managed to form that C-N bond.
To do this, they used the rather simple approach of using an excess of base, deprotonating the most acidic center - the secondary amine. However, that excess allowed a further deprotonation to take place, this time at the proximal benzyl methylene group, forming a chelated dianion bridged by two lithium atoms (figure 3). With the desired atomic partners now tethered together, a little iodine was then able to oxidise this system into a fully-fledged C-N bond, completing the lyconadin A skeleton. Sarpong thoroughly analysed this reaction, adding D2 O (‘heavy water’) to the metalated species and examining the site of deuterium incorporation - leading to their proposed mechanism.
With only one further step required (dimethylation with sodium ethanethiolate), this synthesis represents a very succinct synthesis of a potentially medicinally important target.
References
1 A Bisai, S P West and R Sarpong, J. Am. Chem. Soc., 2008, 130, 7222 (DOI: 10.1021/ja8028069)
2 S P West et al , J. Am. Chem. Soc., 2009, DOI: 10.1021/ja903868n
3 T Cohen and G L Deets, J. Am. Chem. Soc., 1972, 94, 932 (DOI: 10.1021/ja00758a035)
4 G Majetich and K Wheless, Tetrahedron, 1995, 51, 7095 (DOI: 10.1016/0040-4020(95)00406-X)










No comments yet