Synechoxanthin
Synechoxanthin
Although their appearance on cartons of dietary supplements is a recent event, science has long known of the existence and power of so-called antioxidants. I put that carefully, as an important group of antioxidants could be more accurately described as sacrificial reductants - species that are easily oxidised and can thereby protect more sensitive functionality from destructive oxidation. It’s perhaps unsurprising that microbiologists often find high concentrations of these sacrificial molecules in cell membranes, providing a metaphorical bulletproof jacket around their metabolic machinery.
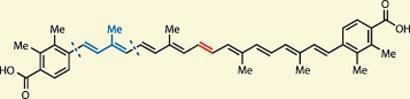
Antioxidants are not only found in human cells (good news), but also in bacterial cell walls (not-so-good news) - and a good example is synechoxanthin. This member of the carotenoid family was found in the cell membrane of a bacterium called Synechococcus PCC 7002 (snappy!), where biological experiments have shown that the carotenoid contributes to its oxidation resistance.1,2 Like all carotenoids, it features an extended (and easy to oxidise) polyene chain; in this case nine alkene units make up it’s nonaene core.
These delicate polyenes represent a considerable synthetic challenge, one particular to those keen on building alkenes. Several general synthetic strategies towards the carotenoids have emerged, but all have issues with a lack of stereoselectivity and inflexibility. Palladium-mediated cross couplings (such as Stille, Suzuki and Heck couplings - see Chemistry World, November 2010, p40) overcome those issues, being more general and importantly, stereospecific, as advocated by seasoned expert, Martin Burke of the University of Illinois at Urbana-Champaign, US.3
Burke’s approach was to reduce the target structure to seven fragments, of which four are identical dienes. Cross coupling these units together seems more than plausible, but would require successive and lengthy functional group transformations to mask and unmask the required halide and halophile coupling partners. However, Burke came up with a cunning solution to that problem in the form of a bis-borylated building block (figure?1), which he uses four times in the synthesis.
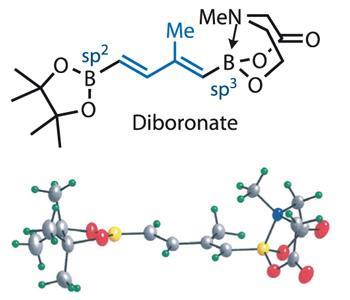
The key to the building block’s design is a pair of masked boronic acids. One is protected in a more traditional sense with pinacol, while the other is bound by Burke’s patented4 N -methyliminodiacetic acid (MIDA), neatly differentiating these moieties. Interestingly, the team isolated this molecule as a stable crystaline solid, and x-ray crystallography reveals the very different hybridisation states of the two boron atoms, and explains their differentiated reactivity.
Modular synthesis
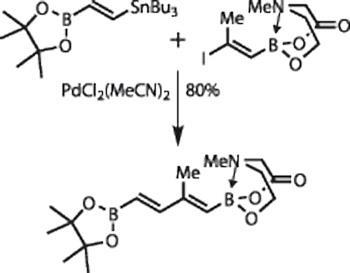
The synthetic action begins with formation of that key intermediate - using, of course, a palladium coupling (figure 2). In this case, a Stille coupling was the reaction of choice, forming the tri-substituted diene in not only excellent yield, but astonishing chemoselectivity, as there are many potential coupling products.
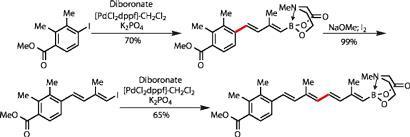
Working directly from this, reaction with an aryl iodide was selective for the pinacol-masked boronic acid, as this protecting group is easily cleaved in situ by potassium phosphate. The resulting product was treated with base and iodine to convert the MIDA borane into a new vinyl iodide. Applying the same coupling conditions as before with more of the diborane unit, that diene was extended to a tetraene, completing almost half of the target in a very respectable yield (figure 3).
All the team needed to do now was stitch two of these moieties together. They did this using trans -1-iodo-2-bromoethylene. Each of the two vinyl halide moieties of this neat little molecule reacted with the MIDA boronate end of two units of the tetraene intermediate, generating the target as its dimethyl ester. A little base (the somewhat unusual potassium trimethylsilanolate, TMSOK) removed both of the protecting groups, finishing the synthesis in eight steps.
Paul Docherty is a science writer and blogger based in Reading, UK
References
1 J E Graham, J T J Lecomte and D A Bryant, J. Nat. Prod., 2008, 71, 1647 (DOI: 10.1021/np800310b)
2 Y Zhu et al, Arch. Biochem. Biophys., 2010, 504, 86
3 S Fujii, S Y Chang, M D Burke, Angew. Chem. Int. Ed., 2011, 50, 7862 (DOI: 10.1002/anie.201102688)
4 M Burke et al, US Patent number 8013203, 2011












No comments yet