
Nickel’s broad catalytic scope is expanded to directly replace the –OH of unactivated alcohols with a carboxylate
Maybe I shouldn’t have favourites, but I do. My favourites change over time without question – as one gets better at something that’s important to me, or another evolves capability from potential. But in the last five years, despite fantastic advances across the board from cobalt to manganese to iron, when it comes to base metal catalysis, nickel trumps them all. I have lauded the properties of nickel before, highlighting its easy oxidative addition into carbon–halogen bonds, or slower β-hydride elimination compared to palladium. But, when it comes down to it, the best thing about nickel is the stuff that it can do but other metals simply can’t. Since Ni(i), Ni(ii) and Ni(iii) oxidation states are all easily accessible, nickel can readily mediate single electron transfers (unlike the heavier platinum group metals), a fact that is widely being exploited in photoredox catalysis. Nickel is clearly head and shoulders above its first row counterparts here, in one of the hottest topics of the moment, but its star qualities do not end there.
Over the last eight years at the Institute of Chemical Research of Catalonia, Spain, Ruben Martin’s group has continually highlighted nickel’s idiosyncrasies, particularly in terms of C–O bond activation and carboxylation reactions using CO2. Martin’s carboxylations of activated and unactivated alkyl halides and pseudohalides is very impressive. Nevertheless, I was always left mildly disappointed by the knowledge that the halides are likely prepared from alcohols, which in turn might have been made by reducing an acid!
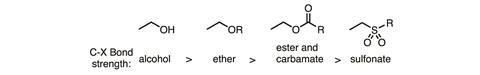
The ideal transformation is clearly to carboxylate alkyl alcohols directly, but their C–O bond strengths exceed that of even the most mildly activated counterparts (figure 1), presenting a formidable challenge. In 2014, Martin and his team set the groundwork, showing that nickel could transform a single allylic acetate into two isomeric allylic acids, with selectivity controlled by ligand selection.1 This was a step in the right direction, but still requires the acetate to be pre-formed.
Now, the team can do the same transformation directly from the alcohol, with CO2 taking a dual role – both providing a carboxylate source and activating the alcohol by forming a carbonic acid derivative, crucially weakening the C–O bond (figure 2).2 To form primary carboxylic acids, the team found that NiBr2•glyme with a phenanthroline ligand, zinc, magnesium chloride and a single equivalent of CO2 gives excellent yields after 16h at 40°C in dimethylformamide. Considering that the conditions are quite similar to those for the transformation of allylic acetates – manganese in place of zinc as the terminal reductant and slightly modified ligands – it is surprising that those conditions give no product at all with alcohols.
A terpyridine ligand is better for generating secondary and tertiary carboxylic acids, almost exclusively forming the more hindered carboxylate. Unfortunately, high yields could only be obtained by switching from bench-stable NiBr2•glyme to the more sensitive nickel cyclooctadiene.
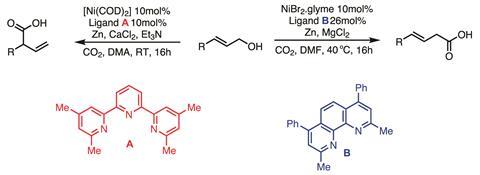
Mechanistically, the reaction is complex and a lot of questions remain unanswered. Treating allylic alcohols under the standard conditions with a preformed Ni(0) bisphenanthroline complex, in the absence of CO2, showed no reactivity at all – indicating that direct oxidative addition of the C–OH bond is highly unlikely and that CO2 plays a rule in activating the alcohol. The team suggests that the presumed carbonic acid intermediate allows nickel insertion into the weakened C–O bond, giving a metal–allyl intermediate. The position of the bond-forming carboxylation seems to be controlled by the catalyst, as it proceeds with near-complete control irrespective of the substrate structure (linear vs branched). The team alludes to distinct η1-Ni(i) complexes with phenanthroline ligands and η1-Ni(ii) complexes with terpyridines, but further investigation is required before the evidence can be considered more than circumstantial.
This work is by no means a holy grail and the reaction of non-activated alcohols is still highly challenging. Catalyst loadings are acceptable at 10%, but ligand loading at 26% is a far cry from ideal when we are looking for increasingly benign reactions, and the necessity of employing Ni(COD)2 to access non-linear products also takes the shine off. Direct carboxylation of allylic alcohols with palladium catalysis and CO2 to give branched products was also reported by Yoshihiro Sato’s group at Hokkaido University, Japan, in 2015,3 although that method requires super-stoichiometric amounts of pyrophoric reagents. What this work demonstrates though, is the increasingly pliable nature of a metal in the first row of the periodic table beyond what was considered possible for many years, and that challenging (stable) functionalities such as alcohols and CO2 – that happen to be ubiquitous, cost effective and relatively benign – are now increasingly under the control of synthetic chemists.
References
1 T Moragas, J Cornella and R Martin, J. Am. Chem. Soc., 2014, 136, 17702 (DOI: 10.1021/ja509077a)
2 M van Gemmeren et al, Angew. Chem.,Int. Ed., 2017, 56, 6558 (DOI: 10.1002/anie.201702857)
3 T Mita, Y Higuchi and Y Sato, Chem. Eur. J., 2015, 21, 16391 (DOI: 10.1002/chem.201503359)













No comments yet