Nanoscale confinement triggers strong base reactivity in process that shuns stoichiometric consumption of resource intensive reagents
US researchers have developed a two-step cycle that inserts carbon dioxide into aromatic hydrocarbons without expensive reagents – an approach that could help turn waste carbon dioxide into valuable carbon feedstock chemicals, such as commodity carboxylic acids and their derivatives.
‘Carbon dioxide is available in relatively pure form at low cost on a very large scale from numerous industrial waste streams but nearly all of it is vented today because we don’t have ways to convert it into valuable products,’ says Matthew Kanan whose team at Stanford University developed the new process.
Methods already exist to form carbon–carbon bonds between hydrocarbons and carbon dioxide. However, these require strong reagents such as organolithiums, zero-valent metals or Lewis acids, which are more valuable than the carboxylate products they produce. What’s more, large amounts of energy are required.
‘We want to make it possible to use carbon dioxide as a carbon source for high-volume chemical synthesis and one of the fundamental challenges for this goal is to eliminate the use of expensive, resource-intensive stoichiometric reagents,’ explains Kanan. To that end, Kanan’s team devised a two-step cycle that converts aromatic hydrocarbons into aromatic esters by simply using carbon dioxide and alcohol as the reagents.
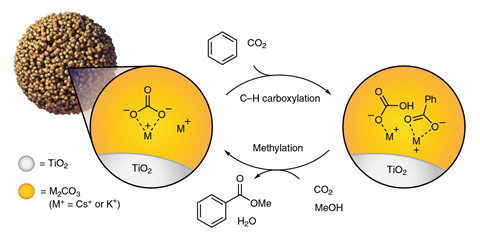
First, the team dispersed a carbonate of caesium or potassium over a titanium oxide mesoporous support. This nanoscale confinement was crucial to disrupt the carbonate’s crystalline structure to turn what is ordinarily a weak base into an amorphous, reactive and potent variant. When heated inside a reactor containing the hydrocarbon benzene and carbon dioxide, the researchers observed that the carbonate deprotonated the benzene in a gas–solid reaction to form an intermediate phenyl anion, which was trapped by carbon dioxide to form benzoate.
In a second step, they exposed the benzoate to a heated stream of carbon dioxide and methanol. This caused the benzoate to methylate and form a methyl ester, which was released from the material and isolated. Meanwhile the carbon dioxide regenerates the carbonate, allowing the cycle to begin again.
‘This is an interesting concept which provides an elegant process to overcome the thermodynamic limitations of direct C–H carboxylations of arenes,’ comments Matthias Beller, an organic catalysis expert at the University of Rostock, Germany. ‘In order to estimate the industrial or practical relevance of the work further studies have to be performed, especially demonstrating the feasability of upscaling.’
‘The major obstacle to turning it into a practical process is that the overall transformation is limited thermodynamically to low equilibrium conversions – only a small fraction of the substrates are converted into product on each cycle,’ explains Kanan. ‘We are working on ways to overcome this limitation by introducing an additional driving force without resorting to non-regenerable reagents.’










No comments yet