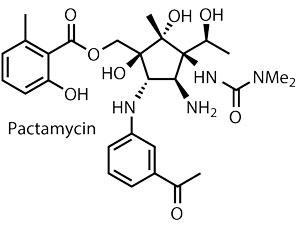
That challenge surrounds analogues of pactamycin, a complex cyclopentane-based target with an exceptionally potent biological profile. The problem with pactamycin lies in that profile – it’s just too active. While its antitumour and antibiotic properties are of huge interest to the medicinal community, its toxicity is holding it back. Genetic engineering has uncovered various analogues of pactamycin, some of which inherit the desirable activity of the parent molecule, but with reduced cytotoxicity. And it is the geneticists who discovered these variants who suggested that organic chemistry is not up to the task of their synthesis.
A small team lead by Jeffrey Johnson of the University of North Carolina at Chapel Hill, aimed to prove those pesky biologists wrong.2 But they had quite a challenge on their hands. Synthetic chemists have had the stereochemically congested structure of pactamycin scribbled on notes marked ‘to do’ for some time, but the first total synthesis fell only recently, in the hands of Stephen Hanessian at the University of Montreal, Canada.3 Pioneering as this work was, the 32 steps required clearly suggested that analogue synthesis might be both time-consuming and inefficient – substantiating the biologists’ bold claims.
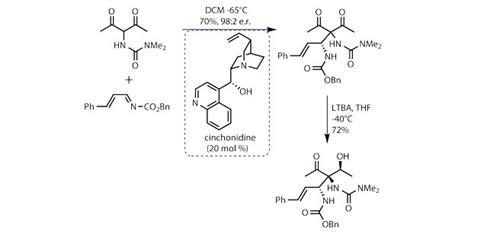
Johnson’s team’s synthesis starts by assembling the carbon skeleton of the cyclopentane, introducing asymmetry at the same time. The first two steps quickly build the 1,3-dicarbonyl starting material. Treating this with a simple imine electrophile and cinchonidine as a catalyst results in an enantioselective Mannich reaction. This delivers the desired alkylation product in both excellent yield and enantiomeric ratio (figure 1).
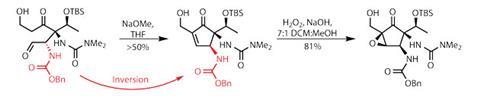
An aldol reaction with formaldehyde adds a ß-hydroxyl group to the remaining ketone. Ozonolysis cleaves the styrene to an aldehyde, and a base-mediated aldol condensation closes the cyclopentene ring (figure 2). The base in this second aldol also ends up inverting the allylic amine to the sterically more favourable (and fortunately correct) configuration.
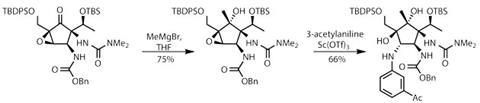
Completing the synthesis required only three further steps to remove all the protecting groups and attach the remaining 2-hydroxy-6-methylbenzoate group. This delivers the target in a remarkable 15 steps – 17 fewer than Hanessian’s pioneering effort. The team may not have begun tackling analogue synthesis just yet, but I feel sure that this flexible and efficient strategy will once again prove the efficacy of synthetic chemistry.
Paul Docherty is a science writer based in Reading, UK












No comments yet