
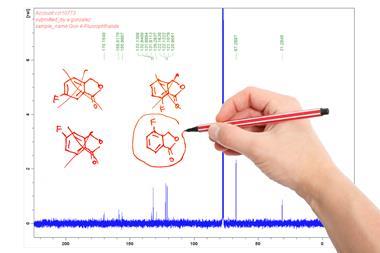
An industry–academia collaboration has led to the development of a tool that combines infrared and proton NMR spectroscopy data to rapidly verify chemical structures.
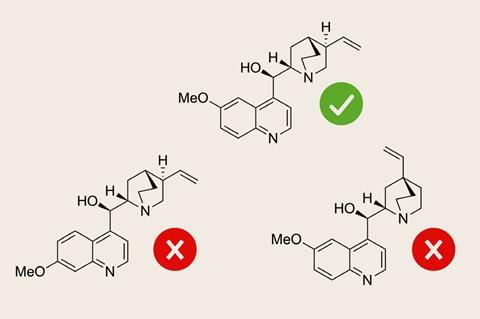
The goal of automated structure verification is to reduce the time organic chemists spend analysing spectra. Rather than solving a structure from the spectra alone, automated structure verification picks the most likely compound from a set of candidates, mirroring how chemists identify reaction products.
Although several tools for this purpose exist, most focus on NMR spectra and are not yet reliable enough to replace human analysis. ‘If an organic chemist looks at a spectrum then they can probably interpret it. But if they’ve got 1000 spectra, which a robot has just churned out, then they’ve got no chance of doing that,’ notes Jonathan Goodman from the University of Cambridge in the UK.
As chemists increasingly adopt automated high-throughput synthesis techniques, the demand for fast and accurate structure verification tools has grown. This need prompted Goodman and his colleagues at Cambridge to partner with researchers from AstraZeneca in Sweden. Together they developed a tool that uses density functional theory calculations to predict proton NMR and infrared spectra from a list of candidate structures. It compares these predicated spectra to the experimental spectra and gives a score that can be used to decide the most likely structure. The researchers found that by combining proton NMR and infrared data, the tool was able to identify the correct structure with a higher confidence than NMR or IR data alone.
‘I think the main advance is the fusion of two quite different spectroscopy methods and showing that this improves the overall structure verification, which I do not think has been demonstrated for structure verification previously,’ comments Kristaps Ermanis, an organic chemist at the University of Nottingham in the UK working on computational NMR spectra prediction.
Untapped potential
One surprising finding from the study was that infrared data alone was almost as informative as proton NMR data. Goodman suggests that with this new tool enabling more structural data to be extracted from infrared spectra, chemists might consider using infrared first, and only turning to NMR if a good structural match isn’t found. ‘Infrared gives really useful information,’ he says. ‘We ought to do more infrared because we can now make better use of the data that it generates.’
‘We probably have a bias towards NMR when we’re thinking about how good a technique is because it’s highly interpretable,’ comments Benji Rowlands, the PhD student who carried out the work. ‘But that doesn’t mean that the infrared spectrum doesn’t contain a lot of information about the structure of the molecule.’
While the tool has advanced automated structure verification, there’s still room for improvement. The team is now exploring machine learning to accelerate and refine spectra prediction.
Ermanis also suggests incorporating other types of structural data in future versions. ‘One could imagine adding mass spectrometry fragmentation data to this as well, since that is a quick and cheap method favoured by industry and is also orthogonal to the other two,’ he says
So how close are we to a future where organic chemists no longer need to manually analyse data without relying on a set of candidate structures?
Goodman believes we’re not there yet. ‘I think it depends how much of a perfectionist you are. Because if you want to put in the spectra, get exactly the right answer every time, I think that’s quite a long way away.’
But Rowlands thinks this is the wrong question. ‘I think that the scenarios in which you would actually want to do that are limited. It’s a much more common workflow that the organic chemist thinks they know what they’ve got and they just need to verify it. From that sort of a verification perspective, I think we’re much closer to having a tool that can reliably tell you whether you’re right or not.’










No comments yet