Karl Collins examines a stealthy strategy for directed C–H activation
Phenols are an incredibly important chemical motif, and are present in many naturally occurring and synthetic molecules. They are also relatively cheap and accessible, making them ideal building blocks. Although phenols have historically been derived from fossil fuels, recent efforts to develop efficient methods to isolate them from biomass mean they may become an increasingly attractive, environmentally sustainable, synthesis feedstock. But if we are to make the most of this resource, it is important we can functionalise phenols in controllable ways.
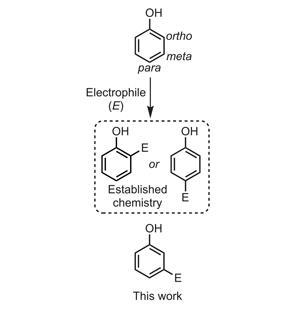
The ortho and para positions of phenols are activated towards electrophiles by the electron-donating hydroxyl group, so we can do chemistry at these positions relatively easily. In contrast, direct meta-functionalisation of these electron-rich aromatics is particularly problematic (figure 1). Some impressive work by Jin-Quan Yu1 of the Scripps Research Institute, US, shows that a directing group on the phenolic alcohol can guide a palladium catalyst to directly activate and functionalise the meta-C–H bond. Unfortunately, installing and removing such a complex directing group independently of the functionalisation step is limiting in terms of cost, time and resources.
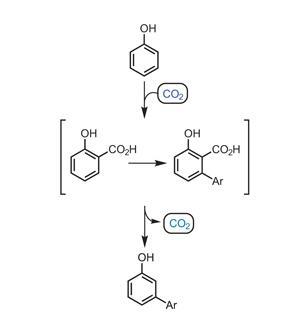
Igor Larrosa and his group at Queen Mary University of London, UK, have made very impressive contributions to the field of C–H activation. The team has now reported a procedure2 that goes some way to addressing the challenge of functionalising phenols at the electronically disfavoured meta position. Using CO2 gas to install a temporary and ‘traceless’ directing group, they can synthesise meta-arylated products from phenols in a one pot process. The strategy (figure 2) is to install a carboxylic acid next to the hydroxyl group using a Kolbe–Schmitt type reaction, and then use this carboxylic acid to direct a C–H activation meta to the hydroxyl substituent (ortho to the acid group), overriding the inherent reactivity of the molecule. Arylation is followed by decarboxylation, which regenerates CO2 to give a meta-functionalised phenol, leaving no trace of the directing group.
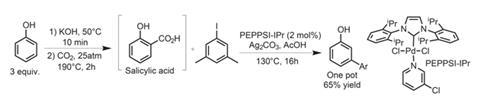
Starting with salicylic acid and an iodoarene, the group first identify efficient conditions for the arylation–decarboxylation process. Using the pyridine-stabilised PEPPSI-IPr pre-catalyst with Ag2CO3 and acetic acid as solvent, they obtain the directing group-free arylated product in 92% yield, after 16 hours at 150°C. They go on to develop a one-pot protocol, starting with the unsubstituted phenol. The carboxylic acid directing group is installed using KOH and a CO2 atmosphere. Adding in the reagents for the arylation–decarboxylation process gives the coupled biaryl in 65% overall yield (figure 3).
The reaction is scalable and tolerates electronically diverse phenols and aryliodides resulting in a pretty impressive substrate scope, though practically it still has some way to go. While this is a one-pot procedure, there are three distinct stages that require practical input, and with reaction temperatures of up to 190°C and pressures of 25atm, this isn’t a reaction anybody will be running on a day-to-day basis. That said, this work is so elegant in many other aspects that I’d still like to try it. Using CO2 gas to install a temporary directing group that can be readily and tracelessly removed is both exemplary and inspiring. Furthermore, this procedure demonstrates an effective strategy to cheat the inherent reactivity of phenols and access the meta-position. There is no doubt that future syntheses will build on this work and expand its utility.
A short investigation shows that the carboxylation process is actually unselective, reacting at both the ortho and para positions. Fortunately, both regioisomers direct functionalisation meta to the phenolic hydroxyl. Hopefully more mechanistic studies will follow, as some insight into the kinetics of the arylation and decarboxylation processes would guide further development. I would also love to know a bit more about the team’s unusual use of a bulky N-heterocyclic carbene ligand in C–H activation chemistry.
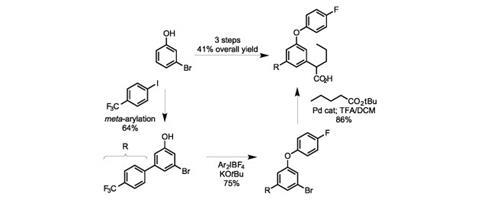
The team completes this study with an efficient three-step synthesis of a gamma-secretase inhibitor, starting from 3-bromo phenol in 41% overall yield, far surpassing the eight steps and 6% overall yield of the patented process. While this reaction isn’t the most practical solution for meta-functionalising phenols, it is an impressive piece of work and I can’t wait to see what Larrosa and his colleagues come up with next.
Karl Collins (@karlDcollins) is a research associate at the University of Münster, Germany, and blogs at A retrosynthetic life











No comments yet