
A steric shield – a simple bulky cation – makes it possible to attach boron to aromatic rings’ rarely accessed, remote para position. Unlike other para C–H borylation reactions, the method requires neither an elaborate catalyst nor bespoke ligands. ‘I think others will turn to this method because of its ease,’ says Michigan State University’s Robert Maleczka who, together with Milton Smith, led one of the two teams that independently developed the reaction.
‘Putting a boron on [aromatic] molecules is extremely valuable, because it enables a variety of transformations, not the least of which is the Suzuki cross-coupling reaction, which is among the most common ways carbon–carbon bonds are formed – for example in pursuit of drug discovery,’ explains Maleczka.
Transition metal catalysts can attach boron groups onto aromatic rings by directly replacing a hydrogen atom with boron. ‘The vast majority of those have been ortho selective,’ says Robert Phipps from the University of Cambridge, UK, who led the other team. Most arene substituents – carboxylic acids and amides for example – steer catalysts toward their neighbouring carbon–hydrogen bond.
Strategies to access the meta position are rare, but para selective reactions are organic chemistry’s unicorns. Chemists trying to reach this remote position often do so by attaching elaborate directing groups onto the substrate. ‘But to build a structure that reaches that far, it’s going to be quite big,’ says Phipps. ‘If you have to build all that into your molecule to do the reaction, to take it out again – there’s some disadvantages associated with that.’
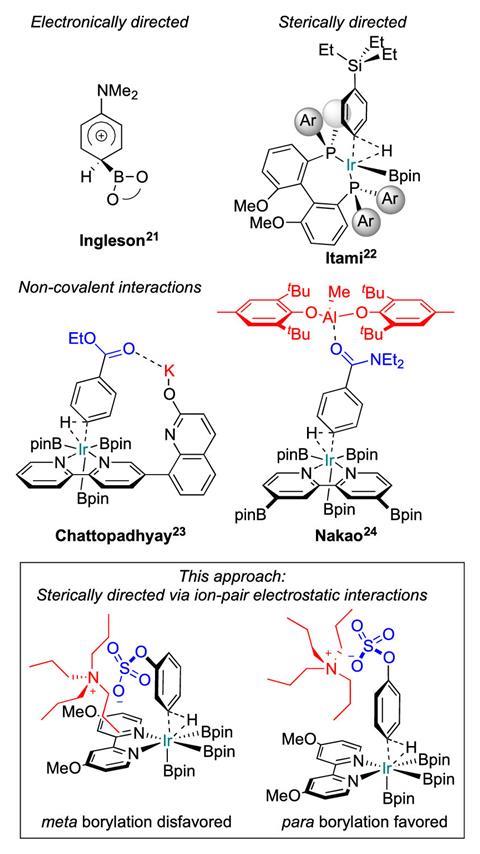
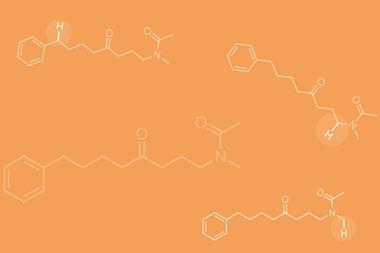
Phipps’ and Maleczka’s iridium-catalysed borylation uses only off-the-shelf reagents and works on aromatics that can be temporarily turned into sulfonate salts: anilines, phenols, benzylamines and benzyl alcohols.1,2 All of the para selectivity comes from the salts’ simple counterion, tetrabutyl ammonium.
‘The cation in the context of these molecules is not something you normally think about, it’s just the solubilising counterion,’ says Phipps. ‘But what we’re trying to do is actually make use of the fact that tetrabutyl ammonium is massive, and we’re using that as the key tool to force the catalyst to go to the para position.’
On guard
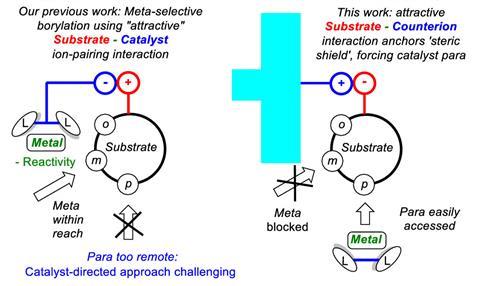
Since the iridium catalyst naturally goes for both meta and para, the cation does not so much direct as discourage it from reacting at the meta position. However, all substrates need to have a 2-substituent – without it, there’s no para selectivity. This is because the steric shield can only protect one side of the ring and one meta position. The 2-substituent disfavours reactions at the other, unguarded meta position.
Nevertheless, the reaction could be useful for cutting the number of steps in natural product syntheses, says Buddhadeb Chattopadhyay, who previously developed a para borylation ligand at the Centre of Bio-Medical Research in India. ‘I am more than sure that this concept would enable many more new things not only for borylation chemistry, but also in other fields of C–H functionalisation chemistry,’ he adds.
Although both Phipps’ and Maleczka’s reaction are built around use of the same counterion concept, each team used slightly different ligands for their catalyst. Phipps and colleagues went for one of the standard bipyridine ligands. Maleczka’s group found a methoxy-substituted bipyridine that improved para selectivity. ‘The difference of many substrates regarding the yield and selectivity of the C–H borylation is remarkable,’ comments Ángeles Fernández-Ibáñez from the University of Amsterdam in the Netherlands, who recently published a para selective olefination.
Why this particular ligand works better than others remains a topic of debate. ‘When [my co-worker Jose Montero Bastidas] has presented his hypotheses on ligand trends, it usually has resulted in a fight, because I’m not so sure we agree on his hypotheses,’ Maleczka laughs.
Maleczka says that ligands can be key ingredients not just for para selectivity but also other unusual reactivity. ‘We’re seeing some results in terms of substitution patterns that in the 20 years this chemistry has been going on nobody has ever seen,’ he says of results that are currently unpublished.
One thing chemists should take away from these studies, says Fernández-Ibáñez, is that ‘we should start looking to non-covalent interactions to control reactivity and selectivity in organic transformations’.
References
1 M T Mihai, B D Williams and R J Phipps, J. Am. Chem. Soc., 2019, 141, 15477 (DOI: 10.1021/jacs.9b07267)
2 J R Montero Bastidas et al, J. Am. Chem. Soc., 2019, 141, 15483 (DOI: 10.1021/jacs.9b08464











No comments yet