
Creating powerful new painkillers is a constant battle against side-effects – particularly addictions. James Mitchell Crow reports
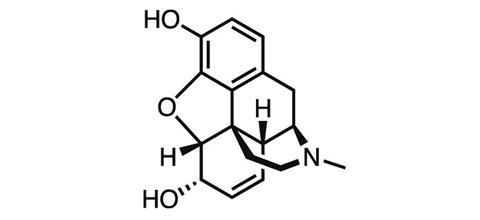
For a chemist whose discovery changed the world, Friedrich Wilhelm Sertürner is a name surprisingly few scientists know. Born in northern Germany in 1783, the accomplished young chemist was working as a pharmacist when in 1804, while studying the extract of the opium poppy, he discovered morphine – one of the first natural products ever isolated.
By discovering a way to isolate a pure analgesic from crude opium resin, Sertürner paved the way for the first dose-controlled medicine for pain. More than 200 years later, morphine is still among the most powerful painkillers we know, and it remains an essential tool in medicine today for controlling severe pain – despite the drug’s notorious side-effects.
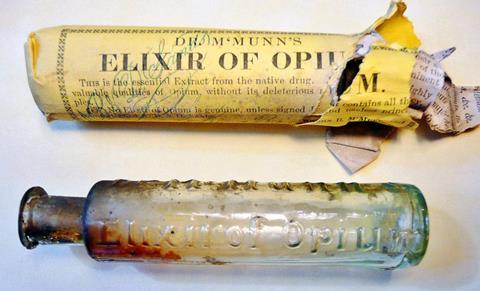
By Sertürner’s day, opium’s addictive properties were already well known. The first known use of the plant dates back to 3400 BC, when the Sumerians of Mesopotamia were already cultivating the opium poppy, which they named the ‘joy plant’.
Unfortunately, opium’s addictive properties carried over into morphine. Sertürner himself may have succumbed to the compound’s insidious grip. He’s known to have tested his discovery on himself, and cotemporary descriptions of the changes to his personality and ‘disturbances of his mood’ in his later life have led many to conclude Sertürner became a morphine addict, perhaps the world’s first.
It wasn’t long after Sertürner’s 1841 death that chemists began looking for opioid compounds that retain morphine’s painkilling properties but which lack its harmful side-effects. These include tolerance – the reduction in the drug’s effect after repeat doses – addiction, constipation and respiratory depression (the side-effect that kills overdose victims). It’s a hunt whose twists and turns have paralleled our evolving understanding of drug–receptor interactions in the body. And a pair of papers published within the last 12 months suggest we are finally closing in on our quarry.
Urgent need
The need for better opioids is urgent. Today, one in five adults is currently in moderate to severe chronic pain. With persistent pain comes a significantly higher chance of unemployment, depression and personal relationship breakdown.
Faced with a lack of effective drugs to help these patients, in the mid-1980s doctors began prescribing powerful opioids such as oxycodone, a close structural relative to morphine, for chronic pain. These compounds’ powerful painkilling effect make them exceptional drugs for short-term control of acute pain, but their side-effects make them a high-risk option for chronic conditions.
‘The first evidence oxycodone was a problem for treating chronic pain came from a Mayo Pain Clinic study published in 1981,’ says Marc Russo, a pain medicine specialist who runs the Hunter Pain Clinic in Newcastle, Australia. ‘It speaks to the power of pain that we accepted that it does the good and the bad – that we haven’t said, “This is not good enough.”’
The resulting health crisis – which has played out in many countries, but is particularly acute in the US – came to be called the ‘opioid epidemic’. By 2014, almost 2 million Americans were classified as addicted to prescription opioids. Nearly 20,000 Americans died that year following a prescription opioid overdose.
In March 2016, the US Centers for Disease Control (CDC) issued new opioid prescription guidelines in a bid to curb the epidemic . But the underlying issue remains: an urgent, unmet need for a safe and effective drug to ease the agony of the millions of people with chronic pain.
Hero or villain?
The first steps along that pathway, taken well over a century ago, didn’t bring much success. One of the first derivatives of morphine ever synthesised was its acetylated form. Diacetylmorphine was first made in 1873, but independently re-synthesised by chemists at Bayer Pharmaceutical Company in 1896, which soon started to sell it under the brand name ‘heroin’ for the supposedly heroic impact it had on users.
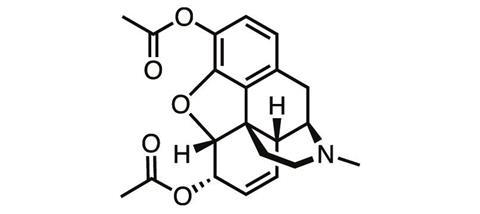
‘Heroin, when it first came out, was touted to be better than morphine because it won’t be addictive,’ says James Zadina, a medicinal chemist at Tulane University and the Department of Veterans Affairs in New Orleans, US. ‘And we know how well that worked out.’ The ‘logic’ behind the argument for heroin’s superiority was that it was more chemically pure than morphine, or more potent, ‘neither of which is relevant’, Zadina adds.
Until the 1930s, we didn’t have any remotely reliable animal tests to be able to assess a drug’s addictive potential. Up to that point, addiction had been thought of as a uniquely human phenomenon. Some of the first indications of addiction’s underlying physiological basis came in experiments on chimpanzees given morphine injections, carried out at the Yale Laboratories of Primate Biology in Florida. Chimps dependent on the drug would start dragging the investigator over to the injecting room for their next hit. The modern ‘conditioned place preference’ animal test looks for similar behaviour in rodents. Rats given a drug with addictive properties will spend significantly more time in the place the drug was administered.
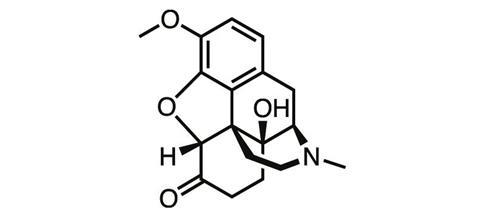
As heroin’s true nature became apparent, chemists went back to the opium poppy, this time trying their luck with thebaine, another natural product of the plant. In a series of oxidation and reduction steps, German chemists at the University of Frankfurt Martin Freund and Edmund Speyer synthesised oxycodone from thebaine in 1916. But the tolerance, addiction and abuse liabilities typical of the opioids remained.
And they still remained in the synthetic opioids such as meperidine (pethidine), discovered in 1939, and fentanyl (1960), which stripped the morphine molecule back to its nitrogen-containing six-membered ‘piperidine’ ring before adding various structural embellishments.
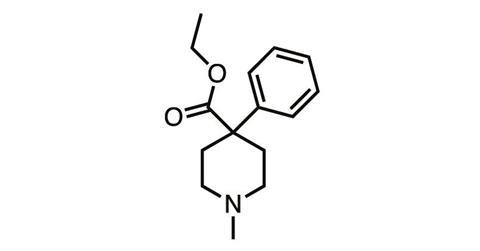
But in parallel with chemical studies, the biology of body–opioid interaction was also being explored – and in the 1970s, it finally seemed like we’d discovered a way to crack the side-effect problem. In 1977, Hans Kosterlitz from the University of Aberdeen (father of Michael Kosterlitz, winner of the 2016 physics Nobel prize) and colleagues showed the body produced its own pain-killing ‘endogenous opioids’, which activated not one but a series of opioid receptor subtypes.
The discovery raised the tantalising possibility that if we could come up with drugs that could selectively target one of the four opioid receptor subtypes – perhaps by using, or adapting, one of the endogenous opioids – we could separate the painkilling effect from the side-effects.
But it didn’t turn out that way. Morphine itself, as well as fentanyl, turned out to be very selective for one of the subtypes, the μ-opioid receptor. Hitting only that subtype seemed to trigger both the good and the bad effects. Hope evaporated. The two sides of opioid behaviour seemed inseparable.
‘I think we thought of receptors as kind of like the drawings: they were pretty, static little circle- and square-shaped holes that get plugged into by little circles and squares,’ Zadina says. ‘We thought any molecule hitting that receptor does the same thing, triggers the same response.’ Now we know the situation is far more complex.
Two decades of work
One of the first indications that the receptor subtype chapter of opioid research was not the end of the story came in 1997. Some 20 years after Kosterlitz’s work, Zadina and his team had looked at known endogenous opioids and the known opioid receptors, and noted something missing: none of the endogenous opioids showed any great selectivity for the μ-opioid receptor that morphine showed such affinity for. None of the endogenous compounds really looked like the natural agonist of that receptor. Delving back into the brain, the team discovered endomorphin-1, the first endogenous opioid whose selectivity for the μ receptor rivalled that of morphine.
And right from the start, endomorphin-1 seemed to elicit a different response in the body from to any other opioid. ‘Even with the original endogenous compound, we could see it was better at analgesia than at conditioned place preference [the animal test of its addictive potential], and better with the respiratory depression,’ Zadina recalls. ‘Which is why I spent the next 20 years making structural analogues from those compounds.’
Zadina worked with three goals in mind. The natural compound, a short peptide, ‘gets chewed up in the blood really quickly’, he says. The team stabilised the structure by forming cyclised analogues, an established technique for fending off the protease enzymes that would otherwise metabolise them in the body.
The other two aims were to maximise analgesia to be as good as morphine or better, while minimising the side-effects, particularly the abuse liability. In late December 2015, Zadina published the culmination of this work, a series of endomorphin analogues that performed so well in animal tests, the team is now gearing up for human clinical trials.
I spent the next 20 years making structural analogues from those compounds
James Zadina, Tulane University
‘What we’ve come upon is a compound that gives very low respiratory depression, much longer duration of analgesia than morphine, and a lot less tolerance,’ Zadina says. But the real focus in Zadina’s lab has been to thoroughly test the addictive properties. One of the most sophisticated abuse potential animal tests is to connect a rat to an IV catheter that will deliver morphine when the animal presses a bar. With morphine in the IV line, the rat will ‘start pushing the bar like crazy’, Zadina says. ‘They don’t significantly increase the bar pressing for our compound. That’s the experiment we think is most compelling, though you never know until you try it in humans.’
What’s behind the promising side-effect profile of Zadina’s lead compound? ‘The old thinking was, any drug that hits that receptor is going to do pretty much the same thing,’ Zadina says. ‘Now we know different structures create different patterns of response.’ The different conformation an agonist adopts within the receptor’s bind pocket triggers different patterns of signalling. ‘The hope is that some set of patterns is going to be better at analgesia, and if you can favour that pathway over the pathways that lead to side-effects you might come out with something better.’
The concept, known as biased agonism, is one of the facets of G-protein-coupled receptor (GPCR) biochemistry for which Robert Lefkowitz at Duke University and Brian Kobilka at Stanford University in the US were awarded the 2012 chemistry Nobel. ‘People are trying to find biased drugs for all the GPCRs, to better control the actions of drugs,’ says Aashish Manglik, a colleague of Kobilka at Stanford.
For opioids, perhaps the most striking result was the work of Laura Bohn who, in collaboration with Lefkowitz, examined the side-effects of morphine in mice lacking β-arrestin, a GPCR regulatory protein. These mice showed enhanced analgesia, and less drug tolerance, constipation and respiratory depression than control mice.
What few people mention, Zadina adds, is that these mice did worse at the ‘conditioned place’ test that assesses addiction, so looking for compounds that simply avoid β-arrestin signalling is unlikely to be the whole answer. ‘My own bias is that it’s not so good to eliminate that,’ he says. ‘You might just have to find the right combination of signalling patterns that the agonist might induce.’
A new hope
Zadina isn’t the only one to look to the brain, rather than the poppy plant, for inspiration for new opioid drug leads. As a grad student in Kobilka’s lab in 2012, Manglik and his colleagues finally cracked the crystal structure of the protein at the heart of it all: the μ-opioid receptor.
‘We were driven primarily by a very basic science interest in how GPCRs function, opioid receptors being a prototypical family,’ explains Manglik, who recently left Kobilka’s lab to set up his own group at Stanford. ‘But the real promise and the dream of GPCR structural biology is to impinge upon drug development,’ he adds. ‘When we first got that picture of the receptor, we decided to team up with Brian Shoichet at the University of California, San Francisco, to see if we could use it to find any new molecules for the mu opioid receptor.’
Manglik, Kobilka, Shoichet and company went on to form a drug discovery super-group . Using Manglik’s crystal structure, the team computationally checked over 3 million compounds for their ability to bind to the μ-opioid receptor, getting 2500 hits. They narrowed this list down to a few dozen that showed the best binding potential, and that had very different chemical structures to existing opioids. ‘We didn’t want to just optimise chemistry that already existed,’ Shoichet says. ‘We wanted to get new chemistry that would confer completely new biology.’ The lead compound was synthesised by Peter Gmeiner’s team at the Friedrich-Alexander University in Erlangen, Germany, then tested at Brian Roth’s lab at University of North Carolina, Chapel Hill, in the US.
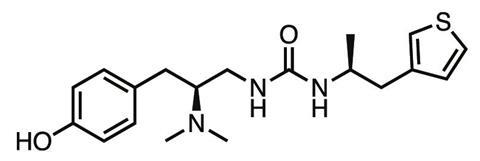
The result was PZM21, a novel compound which in the animal tests carried out so far shows less respiratory depression, constipation, tolerance or addiction than morphine at the equivalent analgesic dose. ‘The molecule is right now a drug lead – we haven’t found a liability that precludes further development,’ Manglik says. ‘For drug development, the probability of failure for a given molecule is pretty high, so we’re continuing to make new versions and find variants that retain the advantageous side-effect profile.’
‘It’s a very exciting study,’ says Zadina. ‘I don’t know if this is going to be the compound,’ he adds, pointing out that so far the team has only published limited data on animal tests of the compound’s addictive properties. ‘But it’s great because it reinforces this idea we should be able to make agonists that separate the good from the bad effects. In the same year, our paper and this paper came out, both reinforcing that idea.’
Manglik agrees. ‘The Zadina group has taken a very rational approach. They have some remarkable properties and some of their data is very exciting.
‘Each of these compounds that has these interesting properties is another way to delve back into the system,’ Manglik adds. Even if these particular biased agonists don’t work out as drug leads in the long run, ‘they’re lead compounds for understanding the biology, to start to hone down into the signalling and really understand what’s going on’. From this understanding, new drug leads will come.
And as Russo points out, this time we’ll want to be very sure before releasing a new opioid into the world. ‘They’ll need to be compared one again the other, clinically evaluated, and not released willy-nilly into physicians’ hands without being certain they’re better than what we have.’
James Mitchell Crow is a science writer based in Melbourne, Australia









No comments yet