
Boron’s chemistry is as much defined by what it isn’t – carbon, or a metal – as by what it is. Recent years have started to fix this misconception, as James Mitchell Crow reports
The periodic table is a symphony of straight lines, of rigid rows and careful columns of precisely positioned elements. But in some depictions of the periodic table, a jagged line slices through that straightness, cutting across the table’s right-hand side like a lightning bolt. To the left of the zigzag lie the metals; to the right, the non-metals. At its apex, cut adrift from the rest of its more metallic family members in group 13 elements, sits boron.
Like any element, boron’s reactivity reflects its position in the periodic table. But boron’s position – like a city atop a tectonic fault line – is more eventful than most.
Like beryllium and lithium, its neighbours to the left, and aluminium, its closest group 13 sibling just below, boron chemistry is characterised by its electropositivity, says Holger Braunschweig from Julius-Maximilians University of Wurzburg in Germany. Yet its position at the pinnacle of group 13 in the periodic table dictates very different behaviour to these neighbouring elements. ‘An electropositive element will give away its electrons and make salts – but boron can’t do that, because it has a very high ionisation potential,’ Braunschweig says. Unable to shed its three valance electrons, boron is forced into covalent bonding instead – but it doesn’t possess enough electrons to form a stable electron octet. The quintessential boron compound is therefore electron deficient: an electrophile and a Lewis acid.
Small-molecule boron chemistry has really come into vogue in the last 10 or 15 years
But over the past decade, boron chemistry has been steadily subverted. A string of boron compounds in exotic bonding states have turned the element’s behaviour on its head. Boron can be coaxed into become a strong nucleophile, a Lewis base, an electron donor. ‘When you make the hypovalent compounds that we make, boron becomes an exceptionally powerful reductant,’ Braunschweig says.
‘At the moment there seems to be an influx of new boron compounds with special reactivity,’ says Rebecca Melen from Cardiff University in the UK. ‘Small-molecule boron chemistry has really come into vogue in the last 10 or 15 years,’ Douglas Stephan from the University of Toronto in Canada agrees. ‘And the landmark in that game is Holger Braunschweig’s work,’ Stephan adds. ‘His 2018 paper, where he activates dinitrogen, is absolutely astounding. It is just going to spur so much activity.’
Being boron
The renaissance in boron chemistry is all the more remarkable given how far main group chemistry had slipped into the doldrums. ‘If you think back 30 years or so, what did we know of boron chemistry, what was boron good for?’ Braunschweig asks. ‘Boron was good for making glasses, and for detergents. Pretty boring, right?’
Today, things are different. Boron is a key functional component of technologies ranging from anticancer drugs (such as the boronic-acid-based multiple myeloma drug Velacade) to OLED displays and solar cells. ‘And then there is the organic synthesis part, Suzuki coupling and everything related to that,’ Braunschweig adds. This transition-metal-catalysed reaction, employing boron as a universal connector for new carbon–carbon bond formation, has becoming a mainstay for synthesising pharmaceuticals and other valuable organic molecules. In 2010, Akira Suzuki shared the Nobel prize for the discovery.

But boron is not just being picked up in fields such as organic chemistry and materials chemistry. Inorganic boron chemistry itself, and main group chemistry in general, is also experiencing a renaissance.
Back in the first half of last century, German inorganic chemist Alfred Stock pioneered boron hydride chemistry. Some of the compounds he created, such as B2H6, seemed to defy basic bonding theory. As it was later discovered, boron was forming cluster compounds to achieve the elusive electron octet. In B2H6, rather than being a direct replica of its carbon analogue ethane, the structure is held together by two bridging hydrogen atoms, chemists established in the late 1940s. Each bridge shares two electrons across three atoms, forming a three-centre, two-electron boron–hydrogen–boron bond that thereby gives each boron atom a complete octet. A far cry from the covalent or ionic bonds we learn about in school.
The discovery showed boron could engage in some weird and wonderful chemistry. But after that time – apart from a few notable discoveries such as the borohydride reducing agents – boron chemistry fell fairly quiet. ‘There hasn’t been very much exploration of main group elements in the grand scheme of things,’ Stephan says. ‘People focussed on organic chemistry, and on transition metal chemistry, and main group chemistry has taken a back seat over the past 50 years.’
One likely reason, Braunschweig suggests, is that boron chemistry is really challenging to do. ‘The compounds are highly air- and moisture-sensitive: they burn in contact with air and whatnot. They are very difficult to make,’ he says. ‘It took a while until this was overcome.’ Gradually, as boron chemistry gained attention for its applications to organic synthesis, culminating in the Suzuki reaction and related transformations, the element was tamed. ‘You had to develop a toolbox to control boron chemistry,’ Braunschweig says.
Developing the necessary know-how to handle boron has underpinned its renaissance, Braunschweig believes. ‘Boron chemistry is always a challenge, so it is very rewarding if you reach your goals,’ Braunschweig says.
New tricks
The latest blossoming of traditional boron chemistry, with the element in its electron-poor three-coordinate state, emerged from Stephan’s labs in 2006. Ordinarily, mixing a Lewis acidic borane with a Lewis base such as a phosphine leads to happy bedfellows. The pair react to form a phosphorus–boron bonded adduct, with phosphorus donating a pair of electrons to the partnership.
But if the alkyl substituents on the boron and phosphorus are bulky enough, the two atoms cannot get close enough to bond. Instead, they form a ‘frustrated Lewis pair’ (FLP).1 ‘What that means is you have a vacant orbital on your Lewis acid – usually boron – and a lone pair on the Lewis base,’ Melen explains. Collectively, the pair can mimic the behaviour of transition metal – with useful reactivity resulting. ‘Transition metals usually have filled and empty orbitals, which act together to activate small molecules such as carbon dioxide, dihydrogen or carbon monoxide,’ Melen says. ‘You can get the same thing with frustrated Lewis pairs – you are just using two different molecules rather than one metal.’
Stephan’s initial focus was to use FLPs as metal-free hydrogenation catalysts. ‘We did simple substrates at the beginning – imines, olefins,’ Stephan recalls. ‘Over the last decade the development of hydrogenation catalysts based on FLPs has become quite advanced – the major advance in the last five years has been the development of really highly asymmetric reduction systems using FLP catalysts,’ he says.
The potential appeal of using FLPs for metal-free reductions is that it would allow pharmaceutical chemists to avoid using transition metal catalysts such as palladium. ‘There are very strict guidelines about the levels of metals you can have at the end of the pharmaceutical process,’ says Melen, for whom developing FLP catalysis forms a major part of her research. ‘Pharma does not use them yet, but I have had several invites to talk at pharmaceutical companies; they are definitely very interested in the idea,’ she adds.
If we can put these tools into the hands of organic chemists, it elevates main group chemistry to a much broader community
Despite significant advance over the past decade, FLPs remain significantly less catalytically active than their transition metal counterparts, so require higher catalyst loadings. FLPs are typically also more sensitive to moisture and less tolerant of different functional groups than transition metals, further negating their advantage. So rather than compete with transition metal catalysis, the most promising path forward is to complement it, Stephan and Melen agree.
One promising possibility is to apply FLPs to activate strong bonds that transition metals activate poorly. ‘Carbon–fluorine bonds are inert to most transition-metal chemistry, but main-group systems seem to be quite active toward them,’ Stephan says. He has used FLPs to do cross-coupling reactions with organic fluorides, he says.
‘If we can put these tools into the hands of organic chemists, it elevates main group chemistry to a much broader community,’ Stephan adds. ‘As main group chemists, we have to make it easy to use, easy to handle, something that is relatively stable and robust.’
Andy Ashley at Imperial College London in the UK is developing water-tolerant FLPs for activating hydrogen, to react with carbon dioxide. The aim is to recycle carbon dioxide emissions into fuels such as methanol. The FLP first activates the hydrogen molecule, Ashley explains. ‘When you activate hydrogen, you get hydride on the boron,’ he says. ‘If you then activate carbon dioxide with the hydride that you get, you are on the first step to reducing carbon dioxide to something useful.’
Again, it is the fact main-group chemistry offers complementary reactivity to transition-metal chemistry that is significant, Ashley says. ‘Transition metals want to bond carbon monoxide because they act as pi donors, whereas frustrated Lewis pairs don’t want to. It is complementary and orthogonal chemistry; it adds more routes to attack the problem.’
Others are incorporating FLPs into metal–organic frameworks, to exploit the specific size cavities inside them to selectively catalyse certain reactions. Meanwhile, Michael Shaver at Manchester University in the UK used FLPs, in combination with small linker molecules, as the basis of a self-healing polymer gel.2 ‘You cut it, then just leave it, and it grows back together,’ Stephan says. ‘If you physically break the Lewis acid and Lewis base apart, they reorient and eventually come back together – it is really cool,’ he says.
Seeking positives
Two electrons short of the full octet, boron compounds are invariably voracious electrophiles. Their reliable Lewis acidity is what makes them so useful in FLPs – but can also be considered a limitation. For example, the organoboron compounds that have become a mainstay of organic synthesis could only be made by electrophilic boron attack on a carbon precursor, limiting the range of boron–carbon bonds that can be formed.
But in 2006, Makoto Yamashita and Kyoko Nozaki – then at the University of Tokyo – flipped 100 years of boron chemistry upside down.3 They created a compound in which boron acts as a nucleophile, opening new pathways to creating organic and inorganic boron-containing compounds. ‘Boron as a nucleophile – this turns the world on its head,’ Stephan says.
The Japanese team’s trick was to find a way to partner boron with lithium, forming a boryllithium salt in which the boron is reduced to a negatively charged ion. Stable lithium salts of boron’s p-block neighbours, such as carbon in methyllithium and nitrogen in lithium amide, are well known. But a stable boron equivalent had never been isolated. Yamashita and Nozaki created the first by reacting lithium with a boron bromide precursor in which the boron atom was wrapped in a bulky nitrogen-based group. Two nitrogen atoms help to stabilise the resulting boron anion, while bulky aromatic sidechains prevent the boron compound from simply dimerising during reduction. The boryllithium species the team produced did indeed behave as a nucleophile, attacking the electropositive carbon atom in aldehydes and alkyl halides, the team showed.
Boron as a nucleophile – this turns the world on its head
Inorganic chemistry has also benefited from the discovery. In 2014, for instance, Braunschweig reacted a boryllithium compound with beryllium chloride to form the first non-cluster bond between boron and beryllium, neighbours on the periodic table.4
But the original boryllithium nucleophile is extremely difficult to handle, Stephan says. He had the idea to create an all-boron FLP, combing a regular boron electrophile with one of the new boron compounds as the nucleophile. ‘I had a student go over to Yamashita’s lab, trying to do boron–boron FLP chemistry - but those things are so hot, the chemistry is really hard to control,’ Stephan says.
Over the subsequent decade, several researchers have stepped in to tame boron nucleophile chemistry. Michael Hill at the University of Bath in the UK has recently synthesised magnesium boryl compounds with relative ease,5 and shown they can borylate aromatic ketones and isocyanates by nucleophilic attack.6
Swing low
Boryllithium compounds offered just a hint of the kind of chemistry that can be done if boron can be coaxed out of its standard +3 oxidation state. Key to the development of low-valent boron compounds has been the intersection of boron chemistry with carbenes, highly reactive carbon species featuring a divalent carbon atom with two unshared valence electrons.
‘Boron’s only stable oxidation state is +3, this is what the textbooks tell us,’ Braunschweig says. In recent years, Braunschweig and others have proved the textbooks very wrong. ‘We have established the boron–boron triple bond, which in a very formal sense would comprise boron in the oxidation state zero,’ he says.7 Braunschweig used bulky, strongly electron-donating N-heterocyclic carbenes to create a compound in which two boron atoms share all six of their available electrons to form the first stable boron–boron triple bond.

In the past few years, a lot of the action has been focused around borylenes, analogues to carbenes and nitrenes, which feature a boron in the +1 oxidation state. Back in 1998, Braunschweig incorporated borylenes into stable transition metal complexes. In 2011, he tried using an N-heterocyclic carbene to generate a metal-free borylene complex, but it was too unstable to isolate and characterize.8 The same year, Guy Bertrand at University of California San Diego made the breakthrough, when he generated an isolable metal free borylene stabilised by two cyclic (alkyl)(amino)carbene (CAAC) ligands.9
As Bertrand noted at the time, the borylene – like boryllithium – behaved as a Lewis base, although its reactivity was rather hampered by the two huge CAAC groups flanking the boron atom. One solution to that problem, it has turned out, was to slim down to a single CAAC ligand – which Braunschweig has put to good use.
‘Our research is curiosity-driven: we get curious about a new class of boron-containing compound and we set ourselves a challenging synthetic goal to make it,’ Braunschweig says. Only after optimising the synthesis, and investigating the new compound’s bonding situation and electronic ground state, does the team start to look into the chemistry, Braunschweig says. ‘What we do now is to use these borylene as metallomimetics.’10
Thanks to their partially filled d-orbitals, transition metals can give and take electrons, and thereby bind and activate a wide array of organic small molecules. FLPs offer one way to recapitulate this behaviour in low-cost, earth-abundant, non-toxic main group elements. But borylenes, with their lone pair and unfilled p orbitals, can do it too, mimicking transition metal behaviour in a single boron atom centre. In 2014, Stephan and Bertrand showed a borylene could bind carbon monoxide.11
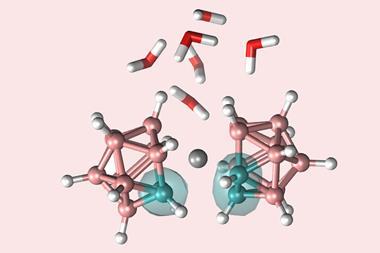
In 2018, Braunschweig put this property to even more spectacular effect when he used it to activate dinitrogen, one of the most stable small molecules known.12 ‘It is one of the real landmarks in main-group chemistry in the last five years at least,’ Stephan says.
The industrial process for activating nitrogen and converting it to ammonia, the Haber–Bosch process, currently accounts for more than 1% of global energy demand. The process is essential for producing the fertiliser that keeps us all fed. ‘When I was a postdoc 40 years ago, the group I was working in was trying to uncover how nitrogenase works and how nature fixes nitrogen,’ says Stephan. Despite another 40 years of intense research, nitrogen fixation is one of those reactions we wish we could do a whole lot better than we can at the moment, he says. ‘Braunschweig’s discovery of a main group system that will capture dinitrogen brings a whole new possibility on how we might approach that.’
It’s the borylene’s electron configuration that is critical to activating dinitrogen. ‘You need a powerful reducing agent that is also a Lewis acid, because you need sigma donation and pi back donation,’ says Braunschweig. ‘The borolyene we use can do both.’ The boron atom can accept sigma donation from dinitrogen into one of its empty orbitals, while simultaneously donating electrons into a dinitrogen antibonding orbital and weakening the nitrogen–nitrogen triple bond.

The dinitrogen study was preceded by computational screening, Braunschweig says. ‘At the time, a few stable borylenes had been reported, none of which were suitable for nitrogen activation,’ Braunschweig says. ‘The postdoc who carried out the study screened possible borylenes. You have to have a subtle balance between stability and reactivity.’ The team isolated an adduct in which a dinitrogen unit, with a nitrogen–nitrogen double bond, was bound by two borylenes. This complex could be further reduced to a nitrogen–nitrogen single bond.
Nobody is predicting a boron-based replacement for the Haber–Bosch process, however. ‘What the discovery does is really put an emphasis on the potential of main-group chemistry in the activation and application of small molecules for catalysis,’ Stephan says.
Braunschweig has that goal clearly in mind. ‘We will try to make these processes – be it the activation of hydrogen, nitrogen, aromatic substrates – reversible,’ he says. ‘Only when these processes are reversible could you dream of making them useful catalytically. I don’t know whether we will achieve it, but we think about it and try to approach it. We see that this is a very interesting and worthwhile target to pursue.’
Boron’s blossoming is unlikely to wither any time soon, as the main-group chemistry revival continues. There are plenty more new compounds still to find, Stephan is sure. ‘Boron is just next door to carbon – there are myriad things you can do with carbon and at this point it seems like there is still lots more boron compounds to discover.’
‘Boron definitely has more surprises awaiting discovery,’ Braunschweig agrees. The Braunschweig group’s latest Science paper, published recently, certainly supports his claim. This time, the team coax dinitrogen gas to form a four-nitrogen chain between two borylene complexes, an unprecedented piece of nitrogen chemistry.13 ‘There are a lot more surprises in store for us,’ Braunschweig says.
James Mitchell Crow is a science writer based in Melbourne, Australia
References
1 D W Stephan, Science, 2016, 354, aaf7229 (DOI: 10.1126/science.aaf7229)
2 M Wang et al, J. Am. Chem. Soc., 2017, 139, 14232 (DOI: 10.1021/jacs.7b07725)
3 Y Segawa, M Yamashita, K Nozaki, Science, 2006, 314, 113 (DOI: 10.1126/science.1131914)
4 T Arnold et al, Chem. Commun., 2015, 51, 737 (DOI: 10.1039/C4CC08519A)
5 A-F Pécharman et al, Nat. Commun., 2017, 8, 15022 (DOI: 10.1038/ncomms15022)
6 A-F Pécharman et al, Organometallics, 2018, 37, 4457 (DOI: 10.1021/acs.organomet.8b00408)
7 H Braunschweig et al, Science, 2012, 336, 1420 (DOI: 10.1126/science.1221138)
8 P Bissinger et al, Angew. Chem. Int. Ed., 2011, 50, 4704 (DOI: 10.1002/anie.201007543)
9 R Kinjo et al, Science, 2011, 333, 610 (DOI: 10.1126/science.1207573)
10 M-A Légaré, C Pranckevicius and H Braunschweig, Chem. Rev., 2019, DOI: 10.1021/acs.chemrev.8b00561
11 F Dahcheh et al, Angew. Chem. Int. Ed., 2014, 53, 13159 (DOI: 10.1002/anie.201408371)
12 M-A Légaré et al, Science, 2018, 359, 896 (DOI: 10.1126/science.aaq1684)
13 M-A Légaré et al, Science, 2019, 363, 1329 (DOI: 10.1126/science.aav9593)







No comments yet