
Study computes diatomic bond orders across the periodic table and reveals new bonding insights
Although bond order is a well-established concept, it has proven hard to define in a way that balances chemical sense and physical rigor. Even simple diatomic molecules can be tricky: chemists have debated whether the attraction in C2 could be called a quadruple bond, for example. To help understand these problems, researchers in the US have introduced a new tool that describes chemical bond orders in terms of contributions from molecular orbitals, as part of a survey of almost 300 diatomic molecules across the periodic table.
Why is it that diatomic compounds, that are so fundamental to chemistry, have not had their bond orders studied comprehensively before?
The new research, by Thomas Manz and Taoyi Chen of New Mexico State University, builds upon a bond order definition Manz proposed in 2017. Most measures of bond order are based on the exchange of electrons between atoms, but popular approximations give inconsistent results depending on the methods used and the way molecular orbitals are defined. Although delocalisation indices in their most accurate form avoid these problems, Manz notes that these are very computationally demanding, and the values they report don’t necessarily reproduce chemical intuition. ‘Why is it that diatomic compounds, that are so fundamental to chemistry, have not had their bond orders studied comprehensively before?’ he asks. ‘In some sense I think [chemists] knew intuitively that they didn’t have a comprehensive method that would be good enough to look at a large number of diatomics, because even in diatomics you encounter a lot of different types of bonding.’ In Manz’s definition, electron exchange is measured but then scaled by the degree of overlap between the atoms. This subtle adjustment avoids strong method dependence and gives bond orders that behave in a way that makes chemical sense, declining as bonds are stretched for example.
In their new study, Manz and Chen calculate bond orders for a wide variety of diatomic molecules, but also provide a way to understand what these bond orders really mean in the form of Bond Order Components Analysis, or Boca. ‘The idea is that with Boca you can have a bond order that’s independent of the orbital representation, but still with the flexibility that it gives you components for any set of orbitals that you choose,’ Manz explains. Manz and Chen compare the exchange of electrons with the exchange of messages in a computer network. The orbitals are whatever channels actually do the exchange, and the bond order components reflect how much exchange occurs through them. As in a computer network, the different components may interact in unfavourable ways – occupying antibonding orbitals can mean they give a positive contribution to the bond order, but lower the contributions of bonding orbitals for an overall negative effect.
Periodic patterns
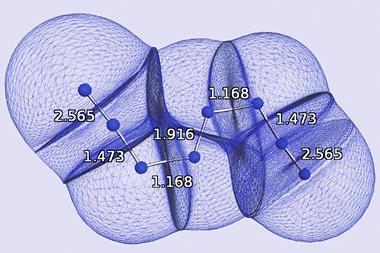
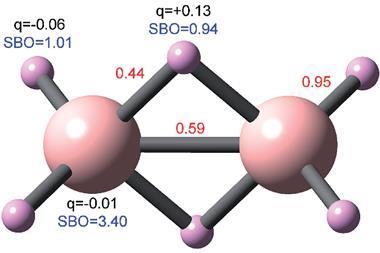
Manz and Chen’s analyses capture a number of the interesting trends and contrasts in the periodic table, such as the greater influence of semi-core electrons on bonding in the alkali metals compared to halogens. Boca also provides a fresh perspective on controversies like the carbon dimer C2. Some interpretations of its electronic structure suggested that it might have a quadruple bond, although the strength and length of the bond suggested it was something between a double and triple bond instead. Rigorous physical arguments exist for the latter, and the apparent disagreement is reflected in the Boca analysis. ‘If people say “we found four bonds”, we can see that there’s four orbitals contributing to the bond but each contribution is less than one, and that’s why the sum of the bond order is less than this number,’ says Chen.
‘Bond order is a central chemical concept and this work offers an interesting approach to calculating it quantum mechanically,’ says Martin Rahm, a researcher in theoretical chemistry at Chalmers University of Technology, Sweden, although he is cautious. ‘How useful is this particular method? This is important to find out because it appears versatile while providing chemically reasonable values. I look forward to future validation through comparison with reactivities of molecules and properties of materials.’ Paul Popelier, a computational chemist at the University of Manchester, UK, praises the breadth of the investigation but is sceptical of the bond order definition itself. ‘Although Manz’s arguments are elaborate they unfortunately contain logical leaps, such as how orbital invariance is achieved or on the need for scaling exchange contributions.’
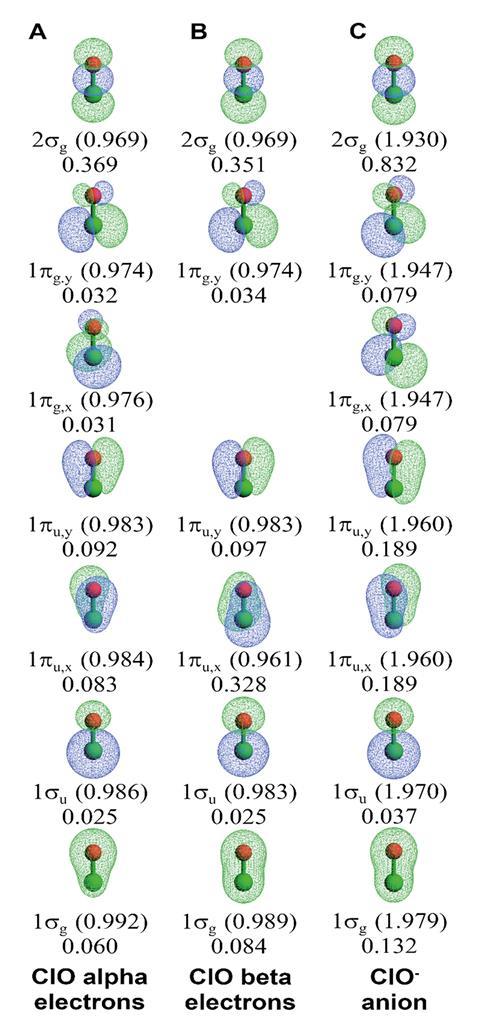
Manz points out that his bond order definition can still behave in unexpected ways, such as in the case of hypochlorite (ClO–); Boca makes these examples easier to explore. ‘You think of the hypochlorite as having just a simple first order bond, and the bond order that we got was in fact 1.74; that’s a huge deviation,’ he explains. Diffuse electrons in anions increase the overlap between the atoms, which is reflected in the total bond order, while Boca showed that a flip in the 1πux orbital compared to the neutral ClO gave it an unexpectedly large contribution. ‘There’s no way we could have expected that was going on. You could probably have honest disagreements about what the correct answer should be, but it’s not obviously wrong since it’s capturing some essence of this.’
References
This article is open access
T Chen and T Manz, RSC Advances, 2019, DOI: 10.1039/c9ra00974d










No comments yet