Japan-based chemists have developed a strategy for activating an anticancer prodrug that takes advantage of elevated levels of endogenous acrolein at tumour sites. This could reduce side effects, which are often serious drawbacks of cancer therapy, by targeting the drug release directly into the cancerous tissue to avoid healthy cells.
Many researchers in the field of chemical biology are combining bioorthogonal chemistry, where reactions take place inside living things without interfering with normal biological processes, with click chemistry to convert prodrugs into active drugs.
The copper-catalysed cycloaddition reaction between acetylene and azide is a well-known example of click chemistry. Katsunori Tanaka from RIKEN and his team were looking for similar reactions that would be applicable to in vivo models when they serendipitously found that aryl azides react with acrolein. Acrolein is a small unsaturated aldehyde which is produced by cancer cells and thus acts as a cancer biomarker. The group found that acrolein is generated in many different types of cancer cells whereas previously it had been difficult to find biomarkers that were general rather than specific to a certain cancer.
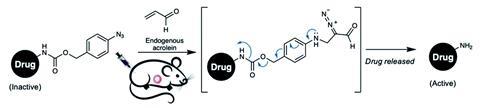
Using the cycloaddition reaction between aryl azides and acrolein as a diagnostic tool, they designed a coumarin-based compound that would release coumarin when it encountered acrolein, leading to an easily detectable increase in fluorescence. The success of this encouraged them to look at using the reaction in a drug release strategy. They replaced the coumarin moiety with mitomycin C, doxorubicin and paclitaxel, all widely available chemotherapeutic agents with toxic side effects, to form prodrugs that would hopefully deliver the active compound to the tumour without causing off-target problems. In vitro studies showed that the mitomycin prodrug had better drug release efficacy and therefore more selective cytotoxicity, and this was tested in vivo. Mouse models showed that the prodrug did indeed stay in the cancer cells where it reacted with the acrolein, releasing the drug into the target area only, inhibiting tumour growth whilst limiting adverse side effects.
‘We make a click-type reaction in the cancer, to treat the cancer; this is our concept,’ says Tanaka. Many current prodrug approaches involve delivering two components to the desired target – traditional click reactions depend on a ligand or enzyme being administered separately. This strategy avoids needing a second component by using endogenous acrolein.
Other researchers working in the prodrug activation area are impressed with the detail that has gone into the work as well as the results. ‘What Tanaka has reported is a very clever and very innovative use of the abnormal metabolism at the tumour site,’ says Binghe Wang of Georgia State University, US. And Floris Rutjes of Radboud University in the Netherlands comments that ‘it is a very interesting approach; to cleave the prodrug in that way is very new.’
References
This article is open access
A R Pradipta et al, Chem. Sci., 2021, 12, 5438 (DOI: 10.1039/d0sc06083f)








No comments yet