Study reveals when, and how, continuum solvent methods fail in computational studies of solvent-phase reaction mechanisms and provide an alternative solvent modelling strategy
Quantum computational modelling can capture physical chemistry in complex solvent-phase reaction mechanisms, new research shows despite recent claims to the contrary.1 The new study introduces a static quantum chemistry scheme with explicit solvent modelling. It can serve as an alternative method to look at solution-phase reaction mechanisms when molecular dynamics simulations are not possible.
‘This project was initiated with the publication of a rather provocative study2 by Daniel Singleton of Texas A&M University,’ explains John Keith of the University of Pittsburgh, US, who led the work. Singleton’s study looked at the complex mechanism for the Morita–Baylis–Hillman reaction; an alkene and aldehyde -coupling reaction catalysed by a tertiary amine. It highlighted the negative effect of using continuum solvent models, which are frequently used in quantum chemistry calculations to account for the solvent in solution-phase reaction mechanism studies. ‘Besides nailing down a complete reaction mechanism, a takeaway from that paper was that all standard quantum chemistry modelling procedures are wrong,’ explains Keith.
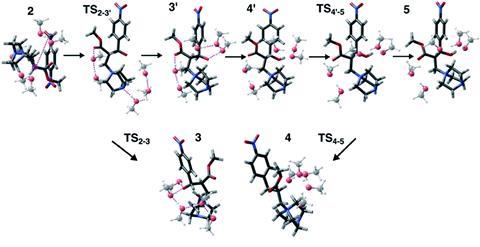
Naturally, this conclusion created a buzz within the modelling community. While some felt that Singleton’s conclusions were too broadly negative, Keith’s group saw them as a rich learning tool. ‘We did not disagree with any conclusions made by Singleton, but we were quite surprised by how poorly computational models using continuum solvent methods were performing,’ he says.
Keith felt that something was fundamentally missing. Maybe the solvent–reactant interactions were significant to the mechanistic pathway? If so, it would be more accurate to include explicit solvent molecules instead of using a continuous model. Validating this hypothesis was not easy: ‘What I first thought would be 6–12 months of work ended up being far more challenging. Fortunately, I had a very talented 1st year PhD student, Yasemin Basdogan, who stayed focused and never quit on the project – or me!’ says Keith.
Their initial plan was to use sophisticated approaches to give solvation energies to a higher accuracy than the ones in Singleton’s study. But the results didn’t add up. It also transpired that a different research team, led by Jeremy Harvey of KU Leuven in Belgium and Raghavan Sunoj of the Indian Institute of Technology Bombay, had similar ideas. Harvey and Sunoj’s study3 reported solvation energies that agreed well with Singleton’s results but they used molecular dynamics methods, which are time-consuming and technically demanding.
No more guessing
Keith changed the focus of his original hypothesis: away from high-level theory and towards understanding why standard modelling methods fail. He showed that continuum solvent models do not describe local solvation effects very well. This can lead to mechanistic steps like proton shuttling and charge transfer being modelled poorly. As an alternative, he developed a strategy that can be carried out by anyone with a general grasp of quantum chemistry.
The first step of the strategy is to identify hypothetical reactant states with different numbers of solvent molecules clustered around them and then globally optimise these. Only a small number of solvent molecules need to be considered: Keith’s team found that using five solvent molecules was adequate for their example. Next, you need to systematically explore the reaction pathway using growing string method calculations to eliminate models with unrealistic barriers to reaction steps that are known to happen experimentally. The growing string method is a simple reaction path and transition-state finding tool. Finally, you should be able to identify a model with a reasonable reaction profile using energies from a trusted level of computation or experimental data if available. An optional extra step is to identify barrier heights between metastable intermediate states for a more complete picture, using the growing string method.
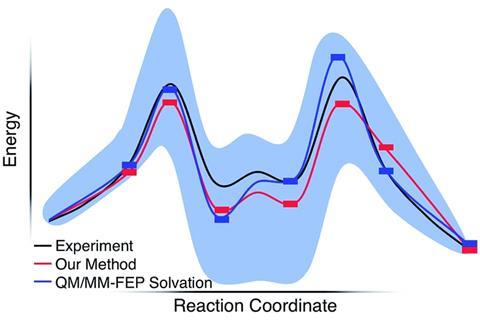
The resulting solvation energies agree with the results of both Singleton and Sunoj despite only explicitly modelling a handful of solvent molecules. Paul Zimmerman, a theoretical chemist from University of Michigan, US, has developed his own code for growing string method calculations. He says Keith’s work puts to bed the past allegations that ‘quantum chemical modelling might not only be nonquantitative, but even qualitatively misleading’. This study shows that ‘physically correct models can be made at reasonable cost, without “guessing”, and chemical simulation can give the right answer for the right reason.’
Keith points out that ‘anyone using this approach should be aware that it is not a substitute for the full and robust simulations that members of the computational dynamics communities have developed for years. This method will only provide an energy profile for a single configuration along a single pathway.’ The user needs to make several trial and error attempts to find an appropriate configuration that agrees with chemical intuition and available experimental data.
References
1. Y Basdogan Y and J A Keith, Chem. Sci., 2018, DOI: 10.1039/c8sc01424h (This article is open access.)
2. R E Plata and D A Singleton, J. Am. Chem. Soc., 2015, 137, 3811 (DOI: 10.1021/ja5111392)
3. Z Liu et al, Phys. Chem. Chem. Phys., 2017, 19, 30647 (DOI: 10.1039/c7cp06508f) (This article is free to access until 30 July 2018.)











No comments yet