
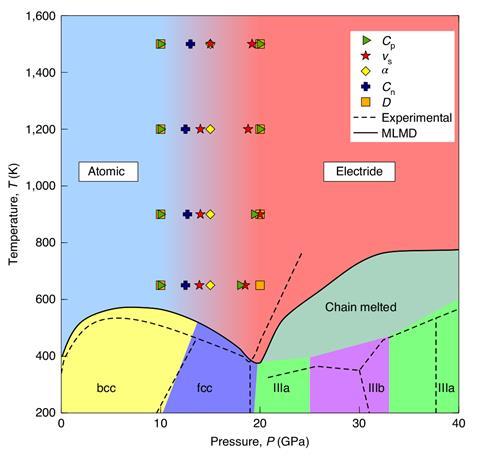
Machine learning-supported simulations have revealed an unusual phase transition in high-pressure liquid potassium. At around 10GPa, the element gradually transitions from a free-electron metal to an electride – a strange, salt-like structure in which the electrons take over the role of the anions.
Little is known about liquid potassium’s behaviour – and that of other alkali metals – at high pressure. This is not that surprising as their high reactivity makes them difficult to work with even in the solid state and at atmospheric pressure. Ab initio molecular dynamics (AIMD) have shown a possible liquid–liquid phase transition in several high-pressure alkali metals indicated by a drop in atomic coordination number. But AIMD calculations can only look at short-range interactions, not at bulk material behaviour, so what exactly was happening remained unclear.
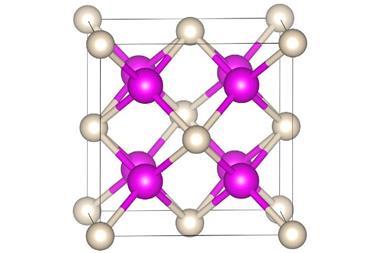
A team of researchers in China, the UK and Italy has now enhanced AIMD with machine learning to simulate the long-range order in high-pressure potassium. They discovered two distinct liquid states: a low-pressure atomic form and a high-pressure electride. In the latter, the electrons act like anions in an ionic salt, settling into the spaces between positively charged potassium nuclei.
The transition from free-electron metal to electride is gradual, the calculations revealed. Starting at around 10GPa, potassium becomes a two-component liquid in which the atomic and electride form coexist until the latter takes over at around 20GPa.
The discovery has also solved the mystery of why potassium’s high-pressure liquid phase is denser than its solid, a face-centred cubic crystal lattice that is the most efficient packing possible for spheres. As the electride’s ions are smaller than atoms they can sit closer together, with the miniscule electrons fitting into the remaining spaces.
The team suggests that other liquid alkali metals should show a similar free electron–electride transition when put under enough pressure.
References
H Zong et al, Nat. Phys., 2021, DOI: 10.1038/s41567-021-01244-w











No comments yet