Model could help drug firms avoid synthetically complex dead ends and speed drug discovery
A method for reliably predicting how well a candidate drug molecule will bind to its target receptor has been long needed by the pharmaceutical industry. It would allow libraries of molecules to be screened on the computer, without having to synthesise them all and test their binding affinities experimentally – and so could speed up the process of drug discovery. A team in the US now claims to have largely cracked this problem.
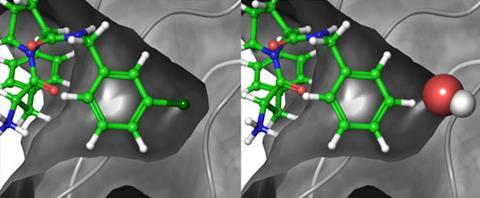
Many drugs work by attaching to the binding site of a protein and blocking or modifying its function. Predicting how well a candidate drug will bind involves calculating the free-energy change in that process. This may be influenced by many factors: electrostatic and van der Waals interactions, hydrogen bonding, entropic effects, changes in solvation and so forth. Such calculations have sometimes been conducted successfully, but it has proved difficult to find methods that will work for many different kinds of ligand–receptor interactions and which don’t require vast amounts of computer power.
The new approach reported by the computational chemistry company Schrödinger, led by Robert Abel, hinges on a better way to calculate the force fields used to model interatomic interactions. It builds on earlier work by co-author William Jorgensen of Yale University and his collaborators, who developed the so-called optimised potentials for liquid simulations (OPLS) force fields in the late 1980s. The researchers have now refined these force fields into a version they call OPLS2.1.
Next generation
The computational scheme also streamlines the algorithms used for sampling different conformations of the receptor–ligand pairing, and reduces the amount of user input needed to run it. Ease of use, they say, is vital for a method that is of real value for drug discovery.
The researchers tested their method for over 200 different ligands binding to eight different receptors, covering a wide range of different systems from cell-cycle enzymes to the blood-clotting agent thrombin. The major driving forces for binding in these examples range from electrostatic to hydrophobic to expulsion of water molecules from the binding site. 138 of the predictions could be compared with experimental measurements and, in most cases, the predicted free energy changes are within 1kcal/mol of the experimentally measured values. About 78% of the compounds predicted to be tight binding were found to be so experimentally and 92% of the molecules predicted to be weaker binding were confirmed by experiment too.
Abel and colleagues don’t pretend that computation will replace experimental studies of new drug lead compounds. But they could narrow the options, for example showing whether it is really worth the effort pursuing a candidate that looks promising but is hard to synthesise. Abel says that the ‘synthesis queue’ is often the major bottleneck in the discovery process.
Other specialists see the work as a potentially useful advance in what is inevitably an incremental process. What drug developers really want to know about binding is the equilibrium association constant K, says John Ladbury of the University of Leeds, UK. But this varies exponentially with the free energy change, so even the small discrepancies that remain can result in orders of magnitude discrepancies in K.
Andrew McCammon at the University of California, San Diego, US, who has developed such computational methods since the 1970s, agrees that hydration changes of the ligand and receptor remain challenging for this kind of calculation, as are the changes of protonation state that accompany the binding of many drugs. ‘Free energy calculations are becoming increasingly reliable and accessible in the world of drug discovery,’ he says, ‘but there is, as always, more work to be done.’











No comments yet