Scientists at the University of Tokyo in Japan have developed a peptoid-based system that can serve as a programmable template for making molecules that inhibit protein–protein interactions (PPIs).1 By overcoming some of the challenges in developing PPI modulators, the system could make it easier for researchers to study these underexplored drug targets.
PPIs are pivotal to a range of physiological processes and are dysregulated in a range of diseases. Consequently, some diseases can and have been treated with molecules that inhibit PPIs. One particular challenge in this field, however, is in making inhibitors that can target PPIs located inside cells, so-called intracellular PPIs. This is because the inhibitor needs to successfully cross the fatty membrane that surrounds cells, and then bind with the target when it gets there.
‘Peptoids [oligomers of N-substituted glycine] have been attracting attention for targeting intracellular PPIs because of their large interaction surface that is suitable for targeting the wide interaction surface area of PPIs and high membrane permeability,’ explains Shinsuke Sando, who co-led the Tokyo team with Jumpei Morimoto. ‘However, there are still only a few peptoids known to inhibit intracellular PPIs.’ One likely reason is because peptoids are very flexible, so only bind weakly to their target, limiting their ability as an inhibitor. To address this in the past, researchers have attached big, bulky groups to the peptoid chain to conformationally constrain it.
Although bulky groups provide stability, only certain groups are suitable. Sando and Morimoto therefore wanted to devise a better strategy for reducing the flexibility in these structures. In a previous study,2 they found that instead of attaching big, bulky groups to nitrogens in the peptoid backbone, they could use smaller and simpler methyl groups to introduce repulsion and constrain its conformation, making oligo-N-substituted alanine (oligo-NSA). This was a promising system, but the PPI inhibitor they designed as a result did not work as a PPI inhbitor inside cells.
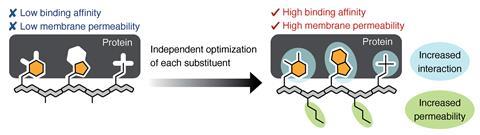
Now, the team has shown that oligo-NSA can act as a template and be tailored for a specific purpose. Along one face of the molecule, users add residues to improve its membrane permeability. And along the opposite face, users add residues to improve the molecule’s affinity for the target protein. The modular nature of oligo-NSAs means binding affinity and membrane permeability can be optimised independently, all the while maintaining the molecule’s backbone shape.
‘This is a really important advance in the field of folded oligomers,’ says peptidomimetics expert Kent Kirshenbaum from New York University in the US. ‘It fills a gap in the ability to pre-organise these molecules, and to allow for predictability in their conformation and complementarity to a protein surface.’
The team used their system to make a molecule that successfully and selectively inhibits the intracellular interaction between proteins MDM2 and p53, a target for cancer therapies.
‘There are similarly reprogrammable template molecules besides oligo-NSA. Macrocyclic peptoids and oligooxopiperazine are such template molecules,’ says Morimoto. ‘The modular reprogramming approach demonstrated in this study can be applied to these other template structures to facilitate the generation of intracellular PPI inhibitors.’
References
1 Y Fukuda et al, Chem. Sci., 2021, DOI: 10.1039/d1sc01560e (This article is open access.)
2 J Morimoto et al, J. Am. Chem. Soc., 2019, 141, 14612 (DOI: 10.1021/jacs.9b04371)








No comments yet