Once considered undruggable, chemists are beginning to grasp protein–protein interactions, according to Ian Le Guillou
Proteins rarely work alone. From tightly bound complexes to passing transitory contacts, interactions between proteins play a central role in the function of the cell. By one estimate, there are over 650,000 protein–protein interactions in the human body. This overwhelming array of interactions represents an almost untapped resource of targets for new therapeutic drugs.
For many decades, these interactions were considered to be ‘undruggable’. High-resolution structures of protein–protein interactions (PPIs) in the 1980s and 90s showed that the interfaces were large, flat featureless surfaces. This was entirely different from the deep pockets found in typical drug targets, such as enzymes and receptors, where small molecules can easily bind.
A typical enzyme binding to its substrate is often taught at school as being like a lock and key. To find a drug to block this interaction, we simply need a new molecule that is sufficiently ‘key-shaped’ to jam the lock. By contrast, our knowledge of PPIs made them seem more like handshakes – two flat surfaces coming together firmly. But our understanding has improved to reveal that it is more complicated than that. There is a secret handshake feel to these interactions, with each side responding to the other and hidden points of contact that are of great significance.
By mutating thousands of individual amino acids at protein–protein interfaces, biochemists found that only a small subset of residues was mostly responsible for binding. These ‘hot spots’ were much more relevant to the strength of the interaction than the size of the interaction surface. This improved understanding made PPIs more tractable as targets, and the need for new, effective drugs certainly made them attractive. However, this is still easier said than done.
I think that protein–protein interactions give you a bit more flexibility
While some natural products approved as drugs, such as taxanes and rapamycin, act by inhibiting PPIs, this was a result of fortune rather than design. Venetoclax, a drug for chronic lymphocytic leukaemia, became the first drug to be approved for targeting a PPI in 2016 and few have followed since.
Although PPIs may no longer be viewed as undruggable, new approaches are needed to deal with this new class of targets. Inhibiting PPIs will need different libraries, assays and perspectives. Given the variety and complexity of PPIs, a range of techniques may be needed to inhibit different targets. As new approaches are developed, many researchers are turning to PPIs to address problems and therapeutic areas that were not previously manageable with traditional small molecule inhibitors.
Selective entry requirements
Most enzymes act on multiple targets in the cell and rely on their protein binding partners to be selective. An inhibitor that blocks the enzyme’s active site would prevent its activity across all of its targets, potentially causing unintended side effects. ‘I think that PPIs give you a bit more flexibility. You can target them to block subsets of enzymes’ activities in a way that’s pretty challenging to do with traditional small molecule enzyme inhibitors,’ says Louise Walport from the Crick Institute in London, UK.
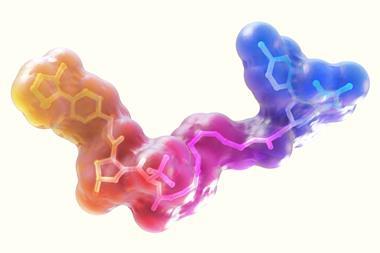
She is studying a group of proteins called arginine deiminases. These are responsible for modifying arginine amino acid residues to remove their charge, which has a knock-on effect for how the proteins behave. The arginine deiminases have a wide range of targets and PPIs may be responsible for managing this. To block these PPIs, Walport screens the arginine deiminases against a library of cyclic peptides. These are molecules of 8–14 amino acid residues in a circular chain.
We can find low nanomolar, sometimes picomolar, binders straight out of a screen
‘I think that PPIs is where peptides have their particular niche because they’re just that bit bigger than small molecules, so they can pick up more small interactions along these featureless surfaces than you can with a small molecule,’ she says. By their cyclic nature there is less entropic cost when they bind to the protein and it helps to make them more stable against being broken down by the body.
The RaPid system, created by Hiroaki Suga from the University of Tokyo in Japan, can generate 1014 different cyclic peptides – more than the number of stars in the universe. The advantage of this approach is that Walport has been able to find ‘low nanomolar, sometimes picomolar, binders straight out of a screen with no modification, not having done any optimisation’.
The challenge, however, is that these molecules are typically too large to enter the cell, meaning that this approach can be used only for extracellular proteins – or a lot of work is needed to adapt the inhibitor to allow it to penetrate the membrane. One notable exception to this is the natural product and immunosuppressant cyclosporine. It is a cyclic peptide of 11 amino acids, but able to enter the cell. ‘No one really understands it. We’d love to be able to make a cyclosporine. It has this kind of flipping mechanism where it can have its hydrophobic face out for a while and then it goes through the membrane and then it flips round – it’s magic,’ says Walport.
A dynamic problem
If proteins often rely on small hot spots for their interactions, however, then it raises the possibility that smaller inhibitors could do the same job, and cyclic peptides with just six amino acid residues would be able to enter the cell relatively easily. This approach, pioneered by Ali Tavassoli from the University of Southampton in the UK, means that he can screen the compound library in live cells.
One of the main advantages of this approach is to study the PPI in a natural environment, says Tavassoli. ‘With assays in vitro, the protein isn’t dynamic – it’s locked in a single state in biochemical buffer. So the hidden pockets and the dynamic nature of the protein, which is one of the core components of its being, is lost.’
It’s completely surprising, but you can make sense of it
Tavassoli studies transcription factors, proteins that can activate or repress genes. By using genetically modified E. coli, Tavassoli can test for compounds that block a PPI responsible for repressing a particular gene. In a life–death assay, the E. coli is placed in a solution containing antibiotics and the PPI is responsible for blocking the gene for antibiotic resistance. If the E. coli survives, then it must mean that the compound has successfully inhibited the PPI.
Through taking advantage of the natural flexibility of the proteins, even very small molecules can interrupt the large interaction surfaces in PPIs. Tavassoli has even found examples where two or three amino acids can block PPIs. One of these even had a 5000Å2 interaction surface – approximately 100 times larger than a dipeptide.
‘It’s completely surprising, but you can make sense of it,’ says Tavassoli. ‘The fact that we’ve got di-peptides, two amino acids, that disrupt this complex – that tells me that these things can’t be working by just getting in between the interacting proteins and disrupting them.’
He suspects that instead the compound is binding to a hidden pocket in one of the proteins that only exists as part of the dynamic transition and prevents the protein from adopting the conformation needed to form the complex.
The compound library that Tavassoli uses, known as Siclopps, generates 3.2 million different cyclic peptides. While the hits may bind a thousand times less tightly than the larger cyclic peptides from the RaPid library, Tavassoli is not too concerned by this. ‘If you want to compete with the substrate of an enzyme, you are going to have to get quite a lot of your inhibitor in there, or it’s going to have to be super-duper potent for it to be effective,’ he explains. ‘But, compare that to the amount of a given protein that’s present in the cell, which is several log-orders less. Potentially you’re going to have to get a lot less of your compound into the cell and equally it doesn’t have to be quite as potent.’
Stabilisation through chemistry
Protein–protein interactions can activate biological pathways as well as repress them, so it’s not always the case that we would want to inhibit an interaction. Luc Brunsveld from Eindhoven University in the Netherlands is using his background in supramolecular chemistry to devise new compounds that can stabilise protein–protein interactions. ‘Stabilisation of protein assemblies is much more like supramolecular thinking than inhibition. Inhibition is classical med chem where you make a molecule that binds to something. But for stabilisation, you talk about bringing multiple things together and the underlying mechanisms,’ he says.
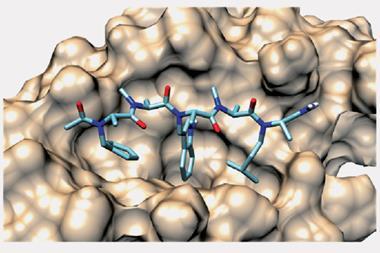
Rather than using a large library of compounds, Brunsveld favours a more structure-based approach. Using the crystal structure of the protein complex, he is looking for opportunities to design a molecule that will stabilise the complex. ‘We want to see, if you can get two proteins together, do they form a novel composite binding pocket for small molecules? That is where the individual proteins don’t have a clear pocket but the coming together of them forms a new binding pocket,’ he says.
Every PPI can be different; you really need to adapt to it
This approach starts with a very small molecule, or fragment, that binds very weakly, and adapting it based on where the fragment binds. There are several techniques that Brunsveld uses to study how they bind to the protein complex, such as soaking them into crystals of the protein complex and using x-ray crystallography to see where it binds or designing the fragments so that they react with the protein in a particular site to form a covalent disulfide bond. ‘For PPI stabilisation, you can’t say there’s a general mechanism; it depends very much on the type of proteins you look at,’ he says.
One of the protein–protein interactions that Brunsveld works on is between 14-3-3, a chaperone protein, and the cystic fibrosis transmembrane ion channel. The ion channel has nine binding sites that can bind to 14-3-3.
This complicated interaction raises questions about how the different binding sites are regulated in the cell and what is the impact of binding to one site over another. For investigating this type of complex, inhibition is unlikely to be successful, says Brunsveld. ‘We see in those multi-valence complexes that inhibition is a big challenge, because as soon as you inhibit one of the nine sites then the others will take over and you will hardly lose affinity [between the two proteins]. But if you specifically stabilise one of the nine binding sites, then that one interaction really becomes dominant and you get huge shifts in the stability of the complex.’
This means that if the different binding sites are important for different processes, then stabilising one will effectively inhibit the others. This provides a very different mechanism for altering biological pathways compared to inhibition. ‘Every PPI can be different; you really need to adapt to it and understand the underlying mechanisms that are acting there,’ says Brunsveld.
Links to cancer
Stabilising the interaction between two proteins isn’t limited to natural binding partners. Bringing any two proteins together offers a powerful tool for influencing biochemical pathways and can even provide a lateral way to inhibit a protein’s activity – by removing the protein altogether.
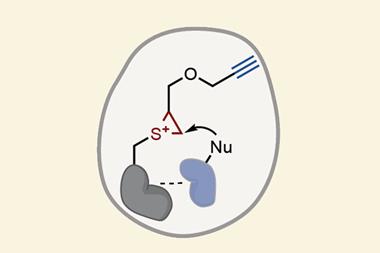
Proteolysis-targeting chimeric molecules – Protacs – are a class of small molecules that have two binding interfaces joined by a linker chain. One end is designed to bind to proteins known as E3 ligases, which tag proteins to mark them for destruction in the cell. If the other end of the Protac is tailored to bind to your protein of interest, the target is brought into contact with the E3 ligase, tagged and destroyed.
Protacs were first suggested 20 years ago, but better understanding of the dynamic nature of these complexes has helped researchers to design better compounds. Alessio Ciulli from the University of Dundee, UK, says that the field initially pictured these complexes being like dumbbells. ‘We thought that were these two heads and then a line in the middle, and so conceptually we didn’t think that the proteins were touching. But of course, the dumbbell is flexible – it can twist and turn. Once these proteins are brought into proximity, then they can form very tight interactions,’ he says.
The binding pocket where the Protac binds is exquisitely conserved
This close-knit protein–protein interaction seems counter-intuitive for two proteins that would normally have little to do with one another. However, it can be used to increase the selectivity for particular proteins. One of Ciulli’s targets is a protein called BRD4, which is very similar to two other proteins, BRD2 and BRD3, making it difficult to target specifically with small molecule inhibitors. Ciulli has developed a Protac that binds to BRD2, BRD3 and BRD4 with the same affinity, and yet the degradation process is highly selective for just BRD4.
‘The reason is that the binding pocket where the [Protac] binds is exquisitely conserved. There’s no difference between the targets. In contrast, the surface around the binding pocket is much less conserved. That’s the region that forms the new protein–protein contacts with the ligase and that’s what gives us specificity,’ says Ciulli. ‘This has provided proof of concept that this is an added advantage of Protacs: that you can discriminate across highly conserved homologues of targets in ways that you can’t do simply with inhibitors.’
Recent research has shown that BRD4 in particular is linked to aggressive forms of prostate cancer. A drug that selectively targets BRD4 but not similar proteins would potentially have fewer side-effects in patients. Just as the proteins are dynamic, so too is the linker chain in the Protac. Ciulli was able to determine the first crystal structure of a ternary Protac complex, and used this to design a cyclic version that more closely resembles the active conformation. ‘This is the first demonstration of this idea of locking the Protac in a bioactive conformation by forming a macrocycle,’ he says. ‘We saw that the compound was extremely active. Interestingly, as a result of that, we lost a lot of binary binding [between the Protac and the target protein]. Despite that, it was as potent as the uncyclised one. So it clearly demonstrated that cyclisation had done something extremely favourable in the process.’
Much like the cyclic peptides used for inhibiting protein–protein interactions, this approach allows the molecule to slip seamlessly between the two proteins, only this time acting as a glue rather than a barrier.
Targeted approaches
Proteins can sometimes be viewed as molecular machines – thousands of molecular cogs spinning to keep the cell alive. However, they are also constantly flexing and being influenced by every other molecule around them. This changing and unpredictable nature makes traditional drug discovery enough of an arduous task. But trying to work around the complex interplay of two proteins at once? It is not hard to see why PPIs were once thought to be undruggable.
But out of this complexity comes opportunity. The approaches for targeting PPIs can provide new tools for delicately manipulating biological pathways in a way that isn’t possible with small molecule inhibitors.
People have had to innovate and invent new things
As our understanding of these interactions has developed, so have the approaches used to manipulate them. Often the techniques are based on ideas from traditional small molecule drug discovery but with a little twist, whether it’s putting the odds in your favour with an astronomical number of potential inhibitors, looking for the hidden sweet spot or designing a molecule based on existing knowledge.
‘I think what really attracts me is the fact that your traditional approaches just haven’t been working against these targets,’ says Tavassoli. ‘And so people have had to innovate and invent new things, which if you just thought about them you would think that they wouldn’t be suitable, and yet it has taken this sort of outside the box thinking to drive the field forward.’
Ian Le Guillou is a science writer based in Paris, France








1 Reader's comment