Why do so many drugs have structural similarities? If you look at many of the best-selling and most-prescribed medicines – like hepatitis C treatment sofosbuvir, antihypertensive lisinopril or asthma drug fluticasone – they have a few things in common: they are densely functionalised, have at least one ring and some unprotected amines or alcohols, and often a scattering of fluorine atoms.
Part of the answer is that drug discovery chemists are limited to a chemical space that can be accessed by known reactions and the available building blocks. They also need to consider biology, so reactions that tolerate basic nitrogen atoms are a must since they feature in almost every single biologically active compound. Reactions using only off-the-shelf reagents that don’t catch fire when they’re exposed to air are also a big plus as there’s often no easy access to specialist equipment like glove boxes.
A 2014 analysis1 showed that only two reactions make up over half of all those used in synthesis to discover and develop drugs: the amide formation and the Suzuki–Miyaura cross coupling. ‘The reason these have contributed so much to medicinal chemistry is that they are the most robust reactions,’ says György Keserű, who leads a medicinal chemistry group at the Hungarian Academy of Sciences.
Ranging from the seemingly simple to potential Nobel-winning transformations, every medicinal chemist has a list of reactions they wished existed. Here’s what this list might look like.
The wish list at a glance
1. Fluorination – Exchanging a specific hydrogen for a fluorine atom in molecules with many functional groups. A reaction that installs a difluoromethyl group would be nice too.
2. Heteroatom alkylation – A reaction that – selectively – attaches an alkyl group onto one heteroatom in rings that have several, such as pyrazoles, triazoles and pyridones.
3. Carbon coupling – A reaction as robust and versatile as traditional cross coupling for stitching together aliphatic carbon atoms – ideally with control of chirality, too. Chemists also want more options for the kinds of molecules they can use as coupling precursors.
4. Making and modifying heterocycles – A reaction to install functional groups – from alkyl to halogen – anywhere on aromatic and aliphatic heterocycles, such as pyridine, piperidine or isoxazole. Reactions that can make completely new heterocycles from scratch would be a bonus.
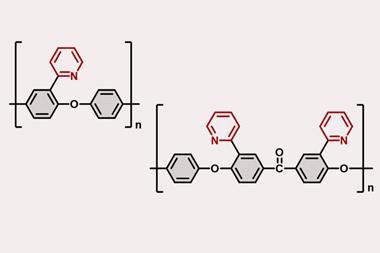
5. Atom swapping – A reaction that can exchange individual atoms selectively, like swapping a carbon for a nitrogen atom in a ring. This chemical version of gene editing could revolutionise drug discovery, but is probably furthest from realisation.
1. Fluorination everywhere, anytime
Perhaps unsurprisingly, a reaction that could convert a C–H into C–F bond is high on the wish list. More than 20% of all commercial pharmaceuticals contain fluorine.2 The antidepressant fluoxetine – better known as Prozac – is a prominent example.

Adding even a single fluorine atom onto a molecule can increase its metabolic stability and lipophilicity. A reaction to quickly fix radioactive fluorine-18 onto molecules could also be a boon for medical positron-emission tomography.
But there’s no straightforward way to replace a specific hydrogen atom with a fluorine once the molecular structure has been assembled, explains Astex’s chief scientific officer David Rees. ‘If you ask a drug discovery chemist when they’ve got their lead molecule: “Can you put a fluorine in each of these positions?” The answer, in general, would be: “I have to go back to the beginning of the synthesis and start with fluorinated starting materials.”’
There are quite a few ways to fluorinate molecules.3 But they all require installing another reactive group – like a tin or boron fragment in aromatics, or a double bond or oxirane in aliphatics – and exchanging it for fluorine. What medicinal chemists are really after is a robust way to directly exchange an H for an F.
But these direct fluorination reactions often struggle with selectivity issues, giving mixtures of regioisomers or over-fluorinating compounds. And since a single fluorine barely changes a molecule’s reactivity or physical properties, removing excess starting material or side-products after a reaction isn’t trivial.
A lot of the same things are true for reactions that install difluoromethyl groups. The CF2H group is a bioisostere of thiols or alcohols – it has similar biological properties to these groups but can lower a drug’s toxicity and increase its bioavailability. ‘We’re more and more designing them into molecules, but the chemistry hasn’t quite caught up,’ says Christopher am Ende, senior principal scientist at Pfizer.
One of the few ways to install a CF2H group uses diethylaminosulfur trifluoride (DAST), a liq uid that turns into explosive bis-(diethylamino)sulfur difluoride when heated. Whether this is better than working with the alternative – gaseous sulfur tetrafluoride, which releases highly corrosive hydrofluoric acid upon contact with moisture – is questionable. ‘Having that nice solid that you can grab off the shelf to install a difluoromethyl group would be a great advance,’ says am Ende.
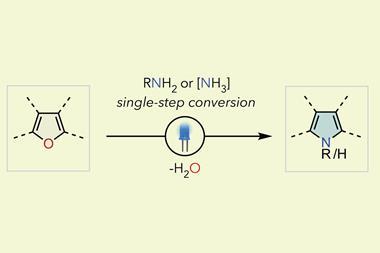
2. Alkylating heteroatoms when there’s several
‘It might be so mundane that most people don’t even want to think about it, but if it could be done that would be incredibly useful,’ says am Ende about the reaction at the top of his personal wish list: pyridone alkylation.
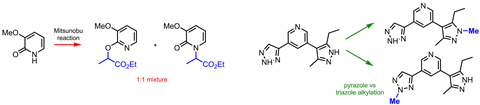
Attaching anything selectively to either the nitrogen or the oxygen in 2- or 4-pyridones is, so far, impossible. Chemists usually end up with wild mixtures of N-alkylpyridones and alkoxypyridines. ‘It’s such a common and simple transformation, I find it hard to believe there isn’t a general fix for this yet with how often pyridones show up in drug discovery,’ am Ende says.
Attempts have been made to figure out the rules behind pyridones’ two-faced reactivity.4,5 But there are so many individual factors, including the pyridone’s substitution pattern, the alkylating agent, the base used for deprotonation, the temperature and even the solvent, it’s still hard to predict – or control – what a reaction will produce.
There are many heterocycles in drug discovery libraries that suffer from similar problems. Pyrazoles and triazoles, for example, have two and three nitrogen atoms, respectively. There’s no reagent combination to selectively modify one of them – they are just too similar in reactivity. And perish the thought of trying to do a selective alkylation on a compound that has several of those heterocycles.
The reason heteroatom alkylation is among medicinal chemists’ most wanted is that these scaffolds make good drugs. The anticancer medications topotecan and irinotecan feature N-alkylated pyridones; O-alkylated pyridones can be found in antimalarial drugs. There is also a whole host of antifungal drugs, like fluconazole, with alkylated triazole units.
3. Powered-up carbon coupling
Cross coupling reactions have enjoyed huge success since their discovery in the 1970s. The reactions that join two – aromatic or other sp2 – carbon atoms together with the help of a palladium catalyst have become essential for building organic scaffolds. But the flat biaryls traditional cross coupling reactions are so good at making are becoming increasingly limited – chemists want their couplings to go 3D.
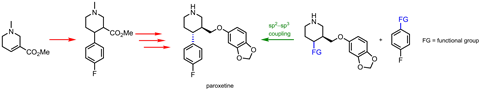
‘Pharmaceuticals are evolving towards higher sp3 carbon content,’ explains organic chemist Mary Watson from the University of Delaware, US. ‘Saturated carbons let you tune a third dimension, which lets you mitigate off-target effects without dramatic changes in other drug-like properties.’ A robust reaction that connects aromatic and aliphatic carbon atoms – or even two aliphatic fragments – is what medicinal chemists are after.
‘Part of the reason people love palladium is that they know what’s going to happen, its oxidation states and redox reactions are well controlled,’ says Watson. But while palladium’s catalytic cycle is perfect for sp2 fragments, it doesn’t do well with aliphatic coupling partners. Undesirable side reactions can takeover, other times the reaction shuts down entirely. What might be needed is a different metal entirely.
Nickel, for example, allows unusual transformations like cross coupling between alkyl amines and alkylzinc halides.6 Other metals might be able to do even more exotic transformations. But ‘how, for example, do you control iron and its wide variation in redox activity?’ Watson asks. ‘What are the right ligands, the right conditions to get it to do what you want it to do?’
But no matter the metal, all cross couplings need pre-functionalisation. Attaching reactive groups onto the coupling partners lets the catalyst know where to join the carbon atoms. Sometimes, for example with pyridines, making these precursors is a big sticking point. So some chemists want to rid themselves entirely of the need to pre-functionalise. The goal is to convert C–H directly into C–C bonds.
The problem is that organic molecules are all about C–H bonds. With so many of them around it’s difficult to tell a catalyst what to do. Currently, reactions rely on workarounds, like the substrate’s inherent reactivity bias or a directing group, to guide the catalyst to a specific C–H bond.
Even if a universal C–H activation reaction becomes a reality one day, ‘I think there will still be times when it will be easier to pre-install a functional group or to take advantage of existing functional groups,’ says Watson. ‘The beauty of organic chemistry is that we created options so we can choose the one that makes the most sense for an application.’
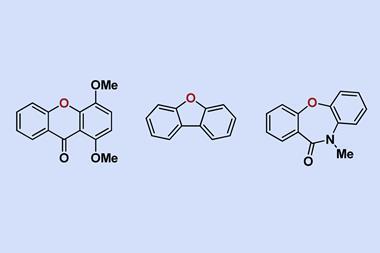
4. Making and editing rings
Heterocycles are medicinal chemists’ bread and butter. Around 60% of all small molecule drugs have a heterocyclic core. By far the most common ones are piperidines, pyridines and pyrrolidines, but there’s also penams, morphinans and isoxazoles among the 25 most frequent nitrogen heterocycles.7
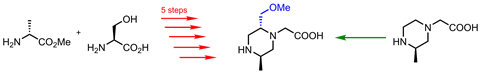
Many small heterocycles are commercially available, but the ones that aren’t can become a major synthesis headache. ‘If you have multi-substituted pyridines, something that may look quite simple on the surface, sometimes you realise that just to make the starting material for your synthesis could take seven, eight, nine steps,’ says am Ende.
Many reactions designed for all-carbon aromatics don’t translate well to heteroaromatics. Fluorinating reagents can react with heterocycles and oxidise rather than fluorinate them. Cross coupling catalysts are often poisoned by heterocycles as they stick to the metal permanently.
There are some ways to install functional groups on aromatic heterocycles late in a synthesis though, like iridium-catalysed borylation. It installs a boron group that can be replaced with a substituent of choice. But figuring out where the reaction might occur – a mix of steric and electronic effects play a role in heteroaromatics – is the subject of entire publications.8 ‘If you could start with pyridine, which is cheap and you can get litres of it, or other simple, abundantly available heterocycles, and you could just go round the ring and selectively install different groups, I’d be out of a job,’ laughs am Ende.
When chemists want an aliphatic heterocycle with unusual substitution patterns, it gets even more complicated. Usually, this means starting from an acyclic precursor. The substituents are installed on the acyclic molecules, which are then painstakingly cyclised later in the synthesis. If the cyclisation fails, it’s back to the beginning.
‘That hurts us because it’s slow,’ says Rees. ‘Frequently, we don’t have time to make those compounds so don’t explore this chemical space even if it might make for really interesting drugs.’
In 2009, chemists at a British biotech company published a paper called Heteroaromatic rings of the future. Hiding behind this poetic title is a list of over 3000 small heterocycles that, although they seem ‘synthetically viable’, had never been made.
Ten years on, a group of French researchers revisited the state of synthesis by looking at the 22 examples the 2009 study had selected as representative.9 Nine of the small bicycles have been synthesised as exact rings. Six have been made as part of larger scaffolds. Another seven have still not been described.
5. Atom swapping
‘For me [it] would be the single most useful new reaction for drug discovery chemistry and might rank alongside Nobel prize winning synthesis,’ says Rees about a carbon–nitrogen swap reaction. This transformation could revolutionise medicinal chemistry if it were possible.

Chemists wouldn’t need to worry any more about how to functionalise heterocycles or how to do a cross coupling in the presence of a few amine groups. Like a chemical version of gene editing, the reaction could take a finished molecule, target a specific carbon atom and exchange it for a nitrogen, oxygen or sulfur.
‘That’s of course unprecedented, and some people don’t like talking about this because they say it’s just unrealistic,’ Rees says. But before Crispr–Cas9, people didn’t think targeted gene editing would be possible either. ‘Science moves very fast and there are a lot of creative people in organic chemistry,’ Rees says. ‘I’m sure it will be done, it’s just a question of when.’
The Baeyer–Villinger oxidation and the Beckmann rearrangement are the two reactions that come closest to a hypothetical atom swap. Both were discovered more than 100 years ago, and both are straightforward: they insert an oxygen or a nitrogen into a cyclic ketone. But both add an atom rather than replacing one.
Acknowledgements
Thank you to Constanze Neumann from the Massachusetts Institute of Technology, and Ben Glasspoole, Kaelyn Wilke and Kenneth Schwieter from Millipore Sigma for helpful discussions.
References
1 D G Brown and J Boström, J. Med. Chem., 2016, 59, 4443 (DOI: 10.1021/acs.jmedchem.5b01409)
2 G K Surya Prakash and F Wang, Chemistry Today, 2012, 30, 30
3 C N Neumann and T Ritter, Angew. Chem. Int. Ed., 2015, 54, 3216 (DOI: 10.1002/anie.201410288)
4 M Breugst and H Mayr, J. Am. Chem. Soc., 2010, 132, 15380 (DOI: 10.1021/ja106962u)
5 M C Torhan, N P Peet and J D Williams, Tetrahedron Lett., 2013, 54, 3926 (DOI: 10.1016/j.tetlet.2013.05.054)
6 S Plunkett et al, J. Am. Chem. Soc., 2019, DOI: 10.1021/jacs.9b00111
7 E Vitaku, D T Smith and J T Njardarson, J. Med. Chem., 2014, 57, 10257 (DOI: 10.1021/jm501100b)
8 M A Larsen and J F Hartwig, J. Am. Chem. Soc., 2014, 136, 4287 (DOI: 10.1021/ja412563e)
9 K Passador, S Thorimbert and C Botuha, Synthesis, 2019, 51, 384 (DOI: 10.1055/s-0`037-1611279)
Further reading
D C Blakemore et al, Nat. Chem., 2018, 10, 383 (DOI: 10.1038/s41557-018-0021-z)
J Boström et al, Nat. Rev. Drug Discov., 2018, 17, 709 (DOI: 10.1038/nrd.2018.217)
K R Campos et al, Science, 2019, 363, eaat0805 (DOI: 10.1126/science.aat0805)
T Cernak et al, Chem. Soc. Rev., 2016, 45, 546 (DOI: 10.1039/C5CS00628G)







1 Reader's comment