Researchers in the UK have quantified the relationship between cation structure and the magnitude of hydrogen-bonding interactions. The findings will help scientists designing new supramolecular systems by predicting how charged species behave in organic solutions.
Non-covalent interactions govern bimolecular recognition and organocatalysis. The ability of cations to form favourable interactions with hydrogen bond acceptors is well known, but there has been no way to rank them relative to one another – until now. Christopher Hunter from the University of Cambridge and his co-workers have turned experimental data into hydrogen-bond donor parameters for the cations guanidinium, primary, tertiary and quaternary ammonium, imidazolium, methylpyridinium, lithium, sodium, potassium, rubidium and caesium.
The team measured the equilibrium constants for the cations when binding to a set of hydrogen bond acceptors and used these to derive the cation’s hydrogen-bond donor parameter. For each cation, they repeated the measurements with different hydrogen bond acceptors in different solvents to check that the parameter was consistent across different systems. They also checked how adding water and different anionic counter ions affected the parameters; both were negligible.
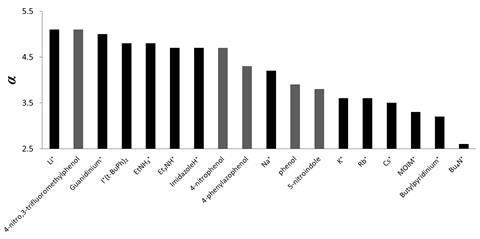
The results for some of the charged cations, like lithium, are unusual. ‘It is surprising that the hydrogen-bonding abilities of charged cations in solution were found to be within the range of neutral hydrogen bond donors, strikingly, some neutral hydrogen bond donors can outcompete fully charged species,’ comments Scott Cockcroft, an expert on the physical chemistry of non-covalent interactions at the University of Edinburgh, UK. ‘The minimal influence of the presence of significant quantities of water and the identity of the anionic counter ions was also surprising.’
Supramolecular chemist Pablo Ballester of ICIQ, Spain, notes ‘the beautiful linear correlation between the experimentally measured free energies of binding and the calculated values using Hunter’s solvation competitive model.’
The new parameters will allow scientists to validate the reliability of existing solvation models. They can also help predict intermolecular interactions in a wide range of contexts, for example, those in water, where ionic interactions are important, and in catalysis, where transition states are generally partially charged.
‘As a supramolecular chemist what you really want to know is if I put an interaction into my system is it going to change my affinity by an order of magnitude or by an undetectably small amount?’ adds Hunter. ‘That’s what this approach allows you to quantify – how big the effect is going to be.’









No comments yet