A weird structure is enough to spark chemists’ interest, even without any obvious use
If you don’t make a habit of reading natural product literature, you could be forgiven for thinking that nature’s molecules are a bit samey. If you’ve only encountered the classes that make their way into undergraduate chemistry courses – amino acids, steroids, maybe a vitamin or two – then you’re probably just used to seeing saturated carbon skeletons with the odd nitrogen or oxygen atom. There’s a lot of that, to be sure, but nature also makes some really crazy stuff – especially when you consider the restrictions of water as solvent and a fairly small number of available reactions. Jawsamycin – named for its collection of tooth-like cyclopropanes – is a great example. At the other end of the complexity spectrum, but perhaps no less impressive, is the ability of certain plants and bacteria to produce ethene gas (from the amino acid methionine). As for functional groups, nature can make far more than you’d think, including terminal alkynes, diazo compounds, alkyl fluorides, isothiocyanates, aryl iodides (human thryoxin has four) and nitroarenes, as well as more heterocycles than you can shake a stick at. The biosynthesis of some of these more unusual classes is not fully understood, but regardless, they are out there!

Structurally interesting molecules attract disproportionate attention from the synthetic community. As bizarre as it may sound, total synthesis chemists would almost always prefer to make a weird-looking molecule than a useful one, as shown by many of the field’s hot targets. The Laurencia family of marine natural products is a good example: a large group of unusual, polyhalogenated molecules that has been enthusiastically studied by the synthetic community. Its best-known member, laurencin, has been made almost 20 times to date, for no reason other the allure of its unnatural-looking structure.
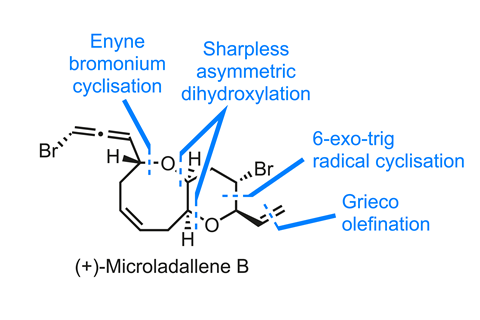
Scott Snyder and his team at the University of Chicago, US, have developed a general approach to several members this popular class.1 Here, I’ll focus on (+)-microladallene B, and pick up the route part way in, with the synthesis of the bromopyran ring. Given Snyder’s interest in bromonium ion chemistry, one could be forgiven for expecting this ring to be forged by a bromoetherification reaction. However, the group actually closes the ring using an unusual diastereoselective 6-exo-trig radical cyclisation under reductive conditions from a geminal dibromide. This unusual tactic was apparently precipitated by failure of the more direct Tsuji-Trost palladium-catalysed allyl substitution approach, which gives the wrong stereochemistry.

A few simple manipulations, including a selenium-mediated Grieco elimination of an alcohol, convert the ester to a vinyl group and install an enyne at the other end of the molecule. This sets everything up for the final key step – the much-anticipated bromonium ion cyclisation. At this point, one might reasonably ask why the team chose to use an ether nucleophile and a programmed ring expansion to get to the 8-membered oxepine ring, rather than the more direct bromonium-induced 8-endo-trig cyclisation. But the latter would be thermodynamically and kinetically difficult, and would produce a slew of different products and isomers. The cyclisation-ring expansion cascade occurs as expected using the somewhat exotic bromodiethylsulfonium bromopentachloroantimonate (BDSB), which was developed by Snyder’s team as a selective electrophilic bromine source. It spits out the target in good yield, but as a mixture of configurations at the bromo-allene.
Congratulations to the team on a successful synthesis of not just (+)-microladallene B but also four more of its cousins!
References
Y A Zhang, N Yaw and S A Snyder, J. Am. Chem. Soc., 2019, 141, 7776 (DOI: 10.1021/jacs.9b01088)











No comments yet