Sometimes synthetic ingenuity can trump nature
As someone with a couple of biomimetic total syntheses under my belt, I can certainly see the appeal of this approach to making molecules. ‘Of course nature knows best,’ the argument goes, ‘it’s been doing this for ages!’.
A few years ago, I asked readers of my blog to pick what they considered the best total synthesis of all time. One of the top contenders was Clayton Heathcock’s legendary synthesis of the daphniphyllum alkaloids, which beautifully exemplifies the advantages of letting nature guide strategy.1 One problem with this approach is that nature can do a lot of things that we just can’t yet – yes it’s clear that terpenoid cancer drug taxol is basically just eight or nine C–H oxidations and a few side chain manipulations from taxadiene, but I’ll wager it’ll be a while before we see it made that way.
Of course, the inverse is also increasingly true, and limiting ourselves to the fairly small number of reactions in nature is a handicap at times, so the best approach necessarily depends on the target. It’s also important to consider why you’re making a molecule, as this influences the strategy. On account of their millenia-long optimisation, biosynthetic pathways tend make a single molecule well, but are usually ill-suited to accessing the range of analogues often sought by medicinal chemists on the hunt for new drugs.
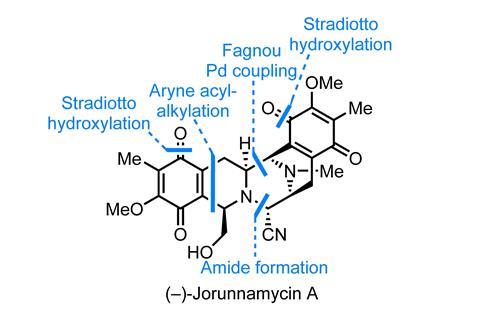
A striking total synthesis of (–)-jorunnamycin A from Brian Stoltz and coworkers at the California Institute of Technology, US, perfectly illustrates the advantages of a non-biometic approach.2 Jorunnamycin is a highly bioactive bis(tetrahydroisoquinoline) – part of a family whose most famous member, ecteinascidin 743, is a marketed cancer drug (Yondelis/trabectidin). Although jorunnamycin and related molecules have been studied a great deal, there remains room in the field for a more efficient, analogue-friendly approach, and this work fits that niche nicely.
First, the team begins by synthesising the bis(isoquinoline) core. Right from the outset, the route is well-suited for introducing structural variation as the team assembles this starting material through a palladium-mediated cross-coupling of two fully assembled isoquinolines. This approach is complementary to methods favoured by other groups, and allows a lot more freedom in how the heterocycles are constructed and what groups can be varied. In this case, the team uses two fairly uncommon methods to assemble the two isoquinolines: an acyl-alkylation of an aryne (a Stoltz group favourite) and a silver-mediated oxime-alkyne cyclisation.
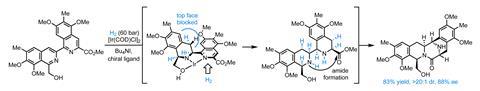
At this point, the team has assembled almost the entire carbon skeleton of the molecule, but surprisingly without any of the necessary stereocentres! Fortunately, this is quickly rectified through an extraordinary hydrogenation reaction that adds four molecules of dihydrogen to the molecule, setting four stereocentres in a single step, and closing the final ring through spontaneous amide formation.
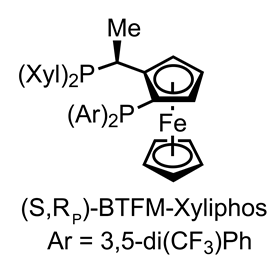
From here, only a few minor adjustments – N-methylation, oxygenation and cyanation – complete the target in just 15 steps. Needless to say, this is an awesome application of asymmetric catalysis and a unique approach to this family of molecules.Although partial hydrogenation of heterocycles is an old reaction, only a handful of asymmetric examples with isoquinones have ever been reported, as the class is particularly problematic. After screening over 60 ligands, the best results were obtained by borrowing from a class of Josiphos-type chiral ferrocenyl phosphine ligands originally developed at Ciba-Geigy for a tricky imine reduction in the synthesis of the blockbuster herbicide Metolachlor.
References
1. C H Heathcock, PNAS, 1996, 93, 14323 (DOI: 10.1073/pnas.93.25.14323)
2. E R Welin et al, Science, 2019, 363, 270 (DOI: 10.1126/science.aav3421)











1 Reader's comment