Pushing the boundaries of C-H activation with Karl Collins
In 1991, Jeremy Knowles of Harvard University in the US wrote that enzyme catalysis is ‘not different, just better’ than small molecule equivalents.1 Nearly 25 years later, this comparison still holds in general. But small molecules are making significant inroads – with reactivity and selectivity approaching levels previously thought unachievable.
One area where enzymes maintain the upper hand is selective C–H functionalisation. Nature’s catalysts are adept at picking out certain of the multiple C–H bonds in complex molecules to react with, guided by their highly organised protein machinery. Christina White from the University of Illinois in Urbana–Champaign, US, is one of the leaders in developing molecular catalysts for this process. Her group’s catalyst systems are beginning to distinguish C–H bonds on the basis of relatively subtle steric and/or electronic differences.
Using a non-haem iron catalyst and relying on the inherent reactivity of substrates, the White group has previously managed to oxidise ‘inert’ C–H bonds within molecules containing multiple potential reactive sites, with fantastic selectivity for a small molecule catalyst. More recently, the White group has begun to overturn substrates’ inherent reactivity – a feat typically associated with enzymes – so that the catalyst controls the site-selection in aliphatic C–H bond oxidation, without recourse to ‘enzyme-like’ catalyst structures.2
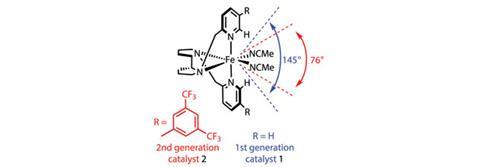
The team has introduced bulky aryl groups bearing two ortho-CF3 substituents onto the pyridine part of the catalyst ligand (figure 1). This modified iron complex maintains its activity, but restricts the trajectory at which substrates can approach the iron centre. The team proposed that this would confer intrinsic selectivity for less hindered C–H bonds.
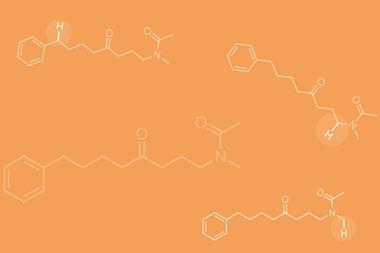
To examine this hypothesis, they compared the selectivity of the original and modified catalysts for a number of substrates (figure 2). Oxidising a linear aliphatic ester shows that the modified catalyst is much more selective for the less hindered secondary carbon over the inherently more electron rich (and hence reactive) tertiary carbon. In contrast, the original catalyst shows no selectivity at all. The modified catalyst can also override the inherent reactivity of a substituted cyclohexane – the original catalyst shows a 2:1 preference for the tertiary C–H site, whereas the modified version selectively oxidises the secondary carbon, giving a 1:2 ratio of products.
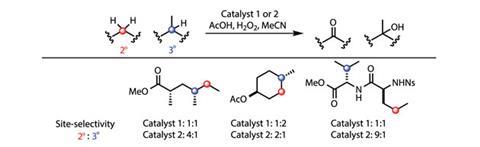
Although the selectivity of these oxidations is notable for the compared reactive sites, if this chemistry were to be scaled up, certain aspects would need to be addressed. The overall selectivity – whether the catalyst acts at either of the potential ‘target’ sites, or somewhere else on the molecule entirely, which has an impact on the reaction yield – in some cases needs improving. One could also argue that the catalyst loading is quite high and the protocol is impractical for large-scale synthesis. Personally though, I consider this work to be more fundamental and inspirational, with the most impressive feature being the combination of the selectivity and substrate tolerance of the catalyst, something not typically seen in enzymes. The high levels of selectivity for oxidising the side chains of a comparably complex dipeptide gives some idea of the reaction’s impressive substrate diversity tolerance (figure 2).
The team has also developed a theoretical model for the catalyst’s selectivity based on substrate structure. The model encompasses both steric and electronic parameters to quantify how susceptible different sites are to oxidation. In line with their hypothesis, the model indicates that steric factors dominate the origin of the selectivity for the modified catalyst, and that the electronics have a negligible influence.
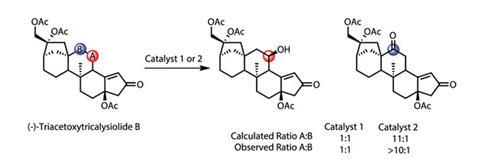
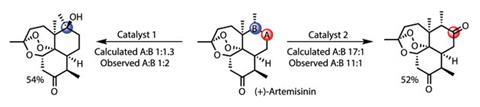
To evaluate this model as applied to complex substrates, and further highlight the broad substrate tolerance of the catalyst, the team oxidises (–)-triacetoxytricalysiolide B, extracted from coffee grounds (figure 3). The theoretical model and experimental results agree well: as predicted, the original catalyst is unselective, while the modified system shows excellent selectivity for the more accessible C–H bond, delivering a remarkable 61% yield. Oxidising the antimalarial drug (+)-artemesinin provides a second beautiful example (figure 4), overriding the inherent reactivity of the molecule. White claims that this level of selectivity has only previously been observed using an enzyme engineered specifically to mediate this single transformation.
Although there is a long way to go until this area of small molecule catalysis can match enzymes’ selectivity and reactivity, this work gives us just a sniff of what may be achievable. More impressively though, it demonstrates a substrate tolerance that any enzyme would be jealous of.
Karl Collins (@karlDcollins) is a research associate at the University of Münster, Germany, and blogs at A retrosynthetic life











No comments yet