



On 19 July 1989, a sudden tragedy altered the lives of the many friends and family of 111 people who died when United Airlines flight 232 crashed at Sioux City, US. Among those killed was John Stille, aged 59.
Stille had spent the last 12 years of his career at Colorado State University, during which time he first published on palladium catalysed carbon–carbon bond formation by coupling between organotin compounds and organic electrophiles. Flight 232 had been en route from Colorado to Illinois, where Stille had taken his first steps in research as a graduate student. The various strands of his doctoral work with Carl Marvel at the University of Illinois in the late 1950s point the way to his later interests. Before all else he was a classical organic chemist – he investigated nitrogen’s inductive electronic effects in condensation reactions of pyridinecarboxaldehyde isomers during his PhD.1 Undoubtedly, his contributions to polymer science find their origin in the training he received under Marvel, and Stille’s interest in organometallic chemistry was also kindled at this time.
Organotitanium chemistry began in 1953, with the first isolation of a compound containing a titanium–carbon bond.2 It was a hot topic. In 1958,3 Marvel and Stille had developed a way to polymerise non-conjugated dienes using i Bu3 Al–TiCl4, one of the emerging family of Ziegler–Natta catalysts whose mechanism was a matter of much debate. Stille was surely interested in the nature of the alkyltitanium catalyst and in complexation of organic ligands by transition metals more generally. In the following years at the University of Iowa, US, he published regularly on the reactivity of organopalladium(ii) species as electrophiles, although concerned himself mostly with polymer chemistry during his early career.
The value of organopalladium chemistry arises from exchange between the metal’s zero and +2 oxidation states. Reduction of Pd(ii) complexes gives electron-rich, nucleophilic Pd(0) species. These readily insert into carbon–halogen (or carbon–OSO2 CF3) bonds, which involves formal oxidation of the metal to Pd(ii). The scope and significance of the oxidative addition process was first explored in the 1960s.4

In the summer of 1977, Stille moved to Colorado, accompanied by postdoc David Milstein. They had found that trans –(phosphine)2 Pdii (R)Cl complexes, made by oxidative addition of a phosphine–bound Pd(0) species into an organic halide, can exchange ligands with an organotin compound. In this transmetalation process, chloride transfers to the more electropositive tin, while an organic ligand migrates to palladium. The Pd(ii) now bears two organic substituents, which rapidly couple together and are eliminated from the metal complex. This reductive elimination regenerates Pd(0) species that can participate in another oxidative addition step that begin the cycle once more.
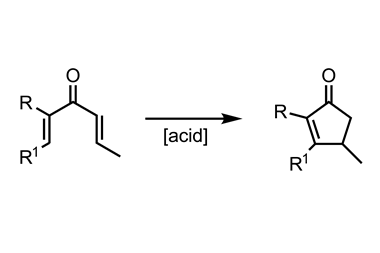
Stille and Milstein were interested in the mechanism of reductive elimination. They used transmetalation from tin to form trans –(phosphine)2 PdII (R)(R1) complexes because of the compatibility of organotin compounds with functional groups like ketones, which would undergo unwanted side reactions with organomagnesium or organolithium reagents (figure 1).5
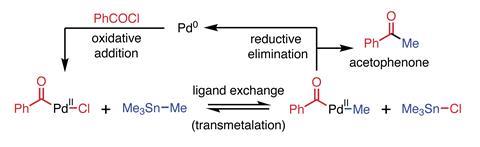
The pair described many high-yielding examples, featuring various different organic groups. It was clear that the reaction would be general, efficient, selective and of great potential in synthesis. Milstein recalls Stille’s joy in recounting that at a conference he had set up the reaction of (n -Bu)4 Sn, benzoyl chloride and the palladium catalyst, the audience watching the apparatus as he lectured. When the solution turned black (from precipitated palladium metal) Stille announced to his excited audience ‘that’s it, the reaction is now over!’
Some pioneering examples of Pd(0) catalysed cross–coupling of organic halides and organotin species had already been reported by groups in the UK6 and Japan.7 Only a few months earlier, Toshihiko Migita and Masanori Kosugi from Gunma University, Japan, had described the same catalytic cycle for a lower-yielding ketone synthesis.8 For this reason the reaction is sometimes referred to as Migita–Kosugi–Stille coupling, but it was the American who worked out the details. Stille interrogated every aspect of the stereospecificity, kinetics and mechanism of all steps in the cross-coupling, adjusting his initial ideas into an understanding that married complexity and elegance.9
His approach was that of the classical organic chemist, using crossover experiments to demonstrate intramolecular trans –cis isomerisation in the diorganopalladium(ii) complex, for example, and monitoring species present in the reaction by 31 P NMR spectroscopy to show that the transmetalation step is rate-limiting.10
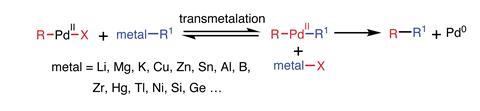
Transmetalation is a hugely important reaction, and extends far beyond tin (figure 2). As reductive elimination is irreversible, even a thermodynamically unfavourable equilibrium for transmetalation is drained towards the organic product R–R1. By tailoring the relative electronegativities of metals using ligands or additives, all sorts of transition metal and main group organometallics can be used. The development of catalytic cross-coupling reactions is a major chapter of late-20th century chemistry, culminating in the 2010 Nobel prize for chemistry. A chapter that owes an immense debt to John Stille.
Acknowledgement
I thank David Milstein for his helpful comments
References
1 C S Marvel and J K Stille, J. Org. Chem., 1957, 22, 1451 (DOI: 10.1021/jo01362a037)
2 D F Herman and W K Nelson, J. Am. Chem. Soc., 1953, 75, 3877 (DOI: 10.1021/ja01112a003)
3 C S Marvel and J K Stille, J. Am. Chem. Soc., 1958, 80, 1740 (DOI: 10.1021/ja01540a059)
4 J A Labinger, Organometallics, 2015, 34, 4784 (DOI: acs.organomet.5b00565)
5 D Milstein and J K Stille, J. Am. Chem. Soc., 1978, 100, 3636 (DOI: 10.1021/ja00479a077)
6 D Azarian et al, J. Organomet. Chem., 1976, 117, C55 (DOI: 10.1016/S0022-328X(00)91902-8)
7 M Kosugi et al, Chem. Lett., 1977, 301 (DOI: 10.1246/cl.1977.301)
8 M Kosugi, Y Shimizu and T Migita, Chem. Lett., 1977, 1423 (DOI: 10.1246/cl.1977.1423)
9 J K Stille, Angew. Chem. Int. Ed. Engl., 1986, 25, 508 (DOI: 10.1002/anie.198605081)
10 J W Labadie and J K Stille, J. Am. Chem. Soc., 1983, 105, 6129 (DOI: 10.1021/ja00357a026)
Additional information












1 Reader's comment