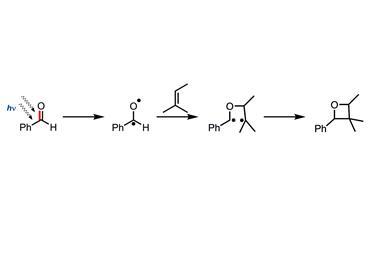



Most organic chemists are familiar with the geminal dialkyl effect, whereby intramolecular ring-forming reactions happen faster when the linking chain between reactive sites includes a tetrahedral carbon atom bearing two alkyl substituents. The corresponding cyclisation with only hydrogen substituents at the same position occurs more slowly (figure 11). The gem-dialkyl effect has often been exploited in synthesis, and it is common to hear the term used interchangeably with ‘Thorpe–Ingold Effect’ – but they are not quite synonymous. Nonetheless, the earliest ideas to explain the rate acceleration came from Jocelyn Thorpe’s laboratory at Imperial College, London, in 1915, where Christopher Ingold was his student.

Born in London in 1872, Jocelyn Thorpe had completed two years of engineering studies before he switched to chemistry on the advice of his father’s friend, Sir Edward Thorpe (no relation).2,3 At the Royal College of Science (one of three institutions from which Imperial College would be founded in 1907) Jocelyn became excited by organic chemistry, which was alive with the possibilities highlighted by a deepening understanding of molecular structure. He hurried to Heidelberg, Germany, to study with Viktor Meyer, before returning to England to work under William Perkin Jr, then as a lecturer in Manchester and Sheffield before his appointment in early 1914 to the new chair of organic chemistry at Imperial. Thorpe explored his many and varied interests, contributing to many theories that persist today – particularly to the early understanding of tautomerism and conjugation in unsaturated systems.

He was also interested in spiro compounds, in which two rings are linked by a common atom. Upon arrival in London he set his assistant, Richard Beesley, to work on the idea that the formation of one ring could be influenced by the bond angles around the linking atom. Beesley began to investigate the spirocycle-forming reactions of 1,1-cyclohexanediacetic acid derivatives, but departed a few months later.

Meanwhile, the Ingold family had relocated from London to the Isle of Wight when their only son, Christopher, was a small boy. His schooling had led to a scholarship at Hartley University College in Southampton, where he studied for a degree in 1913. Ingold’s decision to study chemistry, rather than physics or mathematics which he had preferred hitherto, was influenced by the dynamic teaching of David Boyd, a Scot who had crossed paths with Jocelyn Thorpe in Viktor Meyer’s laboratory.4 Boyd became Ingold’s earliest mentor, a lecturer who demonstrated his subject as one alive with opportunities. It is irresistible to suppose that the young Ingold – later to become one of the most influential chemists of the 20th century with his contributions to the electronic theory of organic reactions – attended Boyd’s lecture to the Scientific Society of Hartley College with the splendid title ‘Discoveries and how to make them’ and perhaps recommended Ingold to join his old acquaintance, Thorpe, in London.

Continuing the work begun by Beesley, Ingold’s first paper with Thorpe soon followed. They proposed that two sterically demanding substituents would move apart, beyond the ideal tetrahedral bond angle of 109.5°, with a corresponding reduction in the angle between the remaining groups. If the two groups being pushed together contain the reacting partners for cyclisation, then this reaction would be faster.5 In forming a spiro[2.5]octane product, Ingold calculated that the compressed angle between the reacting substituents would be ~107.3°, based on the incorrect assumption (common at the time) that cyclohexane was planar, with a bond angle of 120° (figure 2). This intended neat trick of locking the wider bond angle into a cyclohexane ring would cause confusion in the subsequent debate of Thorpe’s central idea – that the bond angles around tetrahedral carbons can be modified by substitution. The delay in acceptance of his suggestion accompanies a series of papers in the 1920s that chronicle Ingold’s increasing acceptance of non-planarity in cycloalkanes. By 1923, he supported the view that rings larger than cycloheptane could adopt configurations whereby the internal bond angles approached 109.5° although maintained that these would be unstable. Importantly, his independent experiments on angle compression effects had now extended to acyclic molecules, allowing more valid comparison of dialkyl- with dihydrogen-substituted systems.6 In the following year, Ingold recognised the possibility of a non-planar configuration for cyclohexane,7 and Thorpe and Ingold acknowledged their earlier mistake in the final paper they would co-author,8 which compiles their full evidence for the ‘valency-deflection’ hypothesis that would soon be known as the ‘Thorpe–Ingold effect’.
We now recognise that the angle compression resulting from dialkyl substitution makes only a negligible contribution to the acceleration of ring-forming reactions. Instead, the dominant effect is to lower the activation energy for cyclisation by increasing the proportion of reactive conformers of the substrate.9 In Ingold’s later words, reported by his biographer, his earliest discoveries were a ‘limited truth’.
References
1 M Newcomb, A G Glenn and W G Williams, J. Org. Chem., 1989, 54, 2675 (DOI: 10.1021/jo00272a042)
2 G A R Kon, J. Chem. Soc. 1941, 444 (DOI: 10.1039/jr9410000444)
3 C K Ingold, Obit. Not. Fellows R. Soc., 1941, 3, 10, 530 (DOI: 10.1098/rsbm.1941.0020)
4 K T Leffek, Sir Christopher Ingold – A Major Prophet of Organic Chemistry, Nova Lion, 1996
5 R M Beesley, C K Ingold and J F Thorpe, J. Chem. Soc. Trans., 1915, 107, 1080 (DOI: 10.1039/ct9150701080)
6 J W Baker and C K Ingold, J. Chem. Soc. Trans., 1923, 123, 122 (10.1039/ct9232300122)
7 C K Ingold, Annu. Rep. Prog. Chem., 1924, 21, 92 (10.1039/ar9242100055)
8 C K Ingold and J F Thorpe, J. Chem. Soc., 1928, 1318 (DOI: 10.1039/jr9280001318)
9 M E Jung, Synlett, 1990, 186 (DOI: 10.1055/s-1990-21028)

![An image showing a single-crystal X-ray derived model of [MH6] complex](https://d2cbg94ubxgsnp.cloudfront.net/Pictures/380x253/4/0/6/501406_full_text__crimmin_161621_859886.jpg)










No comments yet