
Treatments designed to stop amyloid peptide formation seem to cause cognitive impairment rather than prevent it
In yet another setback for Alzheimer’s research, Biogen and Eisai have halted two Phase III clinical trials on the BACE (beta-amyloid cleaving enzyme, or beta-secretase 1) inhibitor elenbecestat in early-stage disease. It is the sixth BACE inhibitor to enter clinical trials only to fail.
‘While there are differences in terms of toxicity among the different drugs, it appears there is a class effect that high-dose BACE inhibition causes cognitive worsening, the opposite of what we are trying to do,’ says Paul Aisen, director of the Alzheimer’s Therapeutic Research Institute at the University of Southern California.
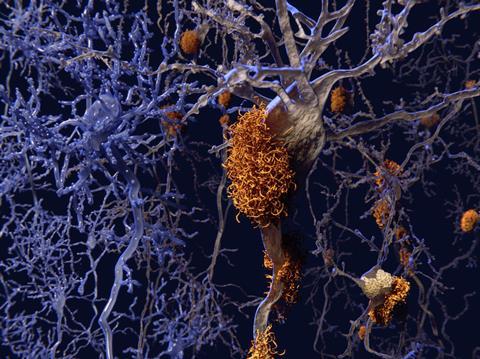
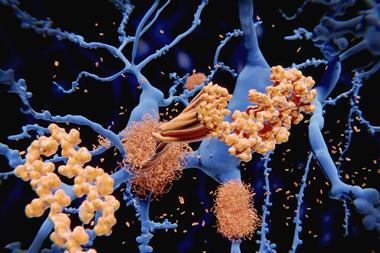
BACE is one of the enzymes responsible for cleaving the plaque-forming amyloid-beta peptide from amyloid precursor protein (APP). Inhibiting BACE or another APP-cleaving enzyme, gamma secretase, should stop amyloid beta production before it can agglomerate into plaques in the brain and cause inflammation.
Aisen does not believe that this is the end for BACE inhibition, however. It may be that the drugs are simply too effective at inhibiting the enzyme, causing the toxic side-effects. ‘The evidence to date suggests that a 70–90% inhibition of the enzyme is too much,’ he says. ‘It is intolerable and interferes with cognitive function.’
He remains optimistic that it could be possible to find a dosing strategy that is both effective and well tolerated. ‘I believe that dosing strategy will aim for 30–50% inhibition of the enzyme,’ he says. This optimism is based on analysis of trial data that suggest that the higher the dose, the worse the cognitive impairment.
BACE inhibition is not the only mechanism targeting amyloid to prove unsuccessful in the clinic – gamma-secretase inhibitors have failed, too, including Lilly’s semagacestat and avagacestat from Bristol-Myers Squibb. Gamma-secretase, like BACE, has multiple substrates in the body, and blocking it can stop its other actions, as well as its activity on APP.
Once you trigger the forest fire of neuroinflammation, you kill 10–100 times more neurons than amyloid plaques and tau tangles kill
Rudolph Tanzi, director of the Genetics and Aging Research Unit at Massachusetts General Hospital in Boston, is not optimistic that either BACE or gamma-secretase inhibition will succeed. He suggests the answer might lie in modulating gamma-secretase, rather than blocking it. As well as early studies using non-steroidal anti-inflammatory drugs (NSAIDs) such as ibuprofen, which proved insufficiently powerful, more recently Tanzi has been developing non-NSAID gamma-secretase modulators with the US National Institutes of Health’s Blueprint Neurotherapeutics Network. They hope to start Phase I trials in the near future.
‘I’m betting on gamma-secretase modulators because then gamma-secretase will still hit all its other substrates,’ Tanzi says. However, he says, it will be important to use it in people who are pre-symptomatic. ‘Once a patient is showing diagnosis, even the earliest stage of mild cognitive impairment, in my opinion that is too late. You really have to hit the brain before it is damaged.’
He compares amyloid plaques to matches, and the tau tangles they form to brush fires, both of which the brain can live with. ‘But once you trigger the forest fire of neuroinflammation, you kill 10–100 times more neurons than plaques and tangles kill,’ he says. ‘That’s when you start showing symptoms, and it’s too late to blow out the match or stomp out the brush fires.
‘I am very optimistic that if we can safely hit neuroinflammation, you will put out the fire, and never underestimate the ability of the forest to grow back. If you stop neuroinflammation, the brain has the chance to recover. We might stabilise, or even improve, Alzheimer’s patients if we can stop neuroinflammation safely.’
Amyloid is still a great target, but you have to hit it five, 10, 15, even 20 years before the symptoms appear. It’s too late once you’re symptomatic
Several companies are looking to address neuroinflammation. The furthest advanced, AZTherapies (Tanzi chairs its scientific advisory board), has a drug in Phase III targeting the gene CD33, which turns microglial cells in the brain into killer cells. Inhibiting this gene could prevent neuroinflammation, and the first data from the trial is due next year. The opposite tack would be to activate another gene, TREM2, which is the ‘off’ switch for this mechanism.
AbbVie and Alector are collaborating on projects targeting TREM2 and CD33, both of which are in Phase I. An alternative approach is being taken by Denali in collaboration with Sanofi, targeting a different protein, RIPK1 (receptor-interacting serine/threonine-protein kinase 1), a signalling protein involved in the tumour necrosis factor receptor pathway. This is also in Phase I.
Results of numerous other Alzheimer’s trials are expected in the next three or four years. These include Eisai–Biogen’s BAN2401, an antibody that prevents amyloid deposition in mouse models, and Aisen is involved in a trial in collaboration with Lilly using an anti-amyloid antibody in clinically normal people, from which early data is due towards the end of 2022.
There is cause for optimism among all the trial failures, Aisen says, not least because of advances in positron emission tomography imaging that allow amyloid and tau levels to be monitored in the brain, and also the ability to measure amyloid peptides in the blood. ‘This gives us a ready tool for monitoring people at any stage, including in a primary prevention approach,’ he says. ‘We also now have tools for monitoring the degree of neurodegeneration with blood tests, we have better clinical trial designs, and we are collaborating and sharing data more effectively. There are many advances that should give us cause for optimism, even in the face of all the frustrating failures.’
But even if a drug can be found, it will need to be dosed in the long term. ‘Any drug for maintaining amyloid levels would have to be taken for decades, the same way people take [statins] for cholesterol,’ Tanzi says. ‘Amyloid is still a great target – the genetics guarantee that – but you have to hit it five, 10, 15, even 20 years before the symptoms appear. It’s too late once you’re symptomatic.’














No comments yet