David MacMillan, a leading light in organocatalysis, takes James Mitchell Crow on a tour of the field

’I hate saying this, but I think it’s true: it was the invention of the word ’organocatalysis’ that really ignited the field,’ says David MacMillan, a pioneer of the discipline. ’There were all these people off doing sporadic things, but once we had a field to work in people could start to recognise it, and it really gelled. It’s amazing how language can do that.’
MacMillan, a Scot by birth now plying his trade at Princeton, the Ivy League university in New Jersey, US, has gone far beyond coining organocatalysis’s catchy name. He has been at the very forefront of this field, and has continued to drive it forward, ever since the area first emerged in 2000.
Organocatalysis - the use of organic molecules to speed up chemical reactions - has captured the imagination of chemists around the world. ’It’s very hot, it’s very exciting, and it’s also intense and competitive,’ says Ben List from the Max-Planck Institute in Mulheim an der Ruhr, Germany, also a major player in the field.
MacMillan first moved to the US in the early 1990s to study for his PhD, and has stayed in the States ever since. ’My first real interactions with catalysis were when I was a postdoc with Dave Evans at Harvard,’ MacMillan recalls. ’My decision to work for Dave wasn’t necessarily based on the fact that he was doing catalysis, but that was something he worked on, and I started to see why that was so important.’
Catalysis in general is a very attractive concept - with just a small sprinkle of catalyst, simple molecules can be speedily clipped together into very complex structures. Not only can each molecule of the catalyst convert many molecules of starting material into product, but the right catalyst will give you only the desired ’enantiomer’; that is, only one of two possible products that are structural mirror images of each other. As MacMillan puts it, ’catalysts can get you to your end product much faster’.
Move over, metal?
New catalysts like organocatalysts always give new types of reactivities, says MacMillan, and ’new reactivity is always a great thing’. Take, for example, the Grubbs catalysts, he says, which catalyses the metathesis reaction, allowing you to link together two molecules (or two ends of the same molecule) through a carbon-carbon (C-C) double bond. ’Before metathesis, many people would have said making C-C double bonds was pretty trivial.’ But this new reaction turned out to be so effective that it is now frequently used as a reliable way to finish the long synthesis of a complex molecule.
Traditionally, catalysis has been the domain of metals. Metals can trigger many powerful transformations. But they aren’t without their drawbacks. ’Most metal-based catalysts, while very successful in many cases, are susceptible to oxygen and moisture,’ says MacMillan. ’As a result you have to work under really inert conditions. Asymmetric catalysis is used in limited form in industry because it’s very tough to produce such incredibly inert conditions on a manufacturing scale.
’We realised the enormous advantages of working with catalysts that are organic in nature, because they’re happy to exist in air. One of my favourite things in the lab is to see my student set up 40 different reactions on the bench top by just scooping out powders into the reaction flasks - not having to use nitrogen or dry solvents [to exclude air and moisture] - and it’s completely fine, they work beautifully.’
Yet no-one is suggesting that metal catalysis will be replaced. ’A metal can activate an organic molecule in a much wider variety of ways than an organic catalyst ever could,’ says Mathew Gaunt, who works on both organocatalysis and metal-based catalysis at the University of Cambridge, UK. ’So organocatalysis will always be complementary to metal catalysis. But, to use the old cliche, it will extend the synthetic chemists’s toolbox of asymmetric reactions quite dramatically.’
Nuts and bolts
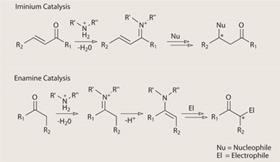
One of the first organocatalytic reactions developed by the MacMillan group was iminium catalysis. This chemistry exploits the fact that carbonyl groups found in aldehydes and ketones react quickly and reversibly with amines, forming an imine. The resulting imine is much more reactive than the initial carbonyl, and will react with a range of suitable partners to give various products. But the real advantage comes when you use an amine that incorporates bulky side chains, so that one face of the intermediate imine is blocked. This shielding means anything reacting with the imine can only approach from the unrestricted side, thereby giving one enantiomer in large excess over the other.
Enantiopure compounds are important because nature is chiral, so any molecules chemists make to interact with nature, such as drugs, herbicides and pesticides, must be the correct enantiomer to trigger the desired response.
’When my group published the paper on [iminium catalysis], I thought it was going to catch on overnight but it didn’t,’ MacMillan recalls. ’But this was just the lag time, and three years later, it just went crazy.’
As MacMillan was discovering imine catalysis, Ben List and Carlos Barbas were working at the Scripps Research Institute, US, to develop enamine organocatalysts. The two approaches are very similar, yet complementary. Both reactions generate activated intermediates from a starting carbonyl and an amine. But where imines are short of electrons and so react with electron-rich coupling partners, enamines have electrons to play with and react with compounds that are electron poor.
Barbas and List’s enamine work was based on the precedent of the Hajos-Parrish reaction, chemistry developed in the 1970s.3 This reaction uses proline as the catalyst, a naturally occurring amino acid that is cheap and readily available in enantiopure form. The Hajos-Parrish reaction is intramolecular - that is, a long chain molecule reacts with itself, effectively biting its own tail to become cyclic. Barbas and List showed that the reaction could be extended to couple two molecules together, and a wide variety of coupling partners have since been shown to react in this way to give products in high yield and with large enantiomeric excess.
Chain reaction
One further benefit of the complementarity of the imine and enamine approaches is that the two processes can be done sequentially in one pot - so-called ’organocascade’ reactions.4
The imine-enamine cascade exploits the fact that, when you react an imine with a nucleophile, the imine functionality is converted to an enamine. If you also have a suitable electrophile in the reaction flask, this will then react with the enamine to give you a doubly substituted product.
Isolating the product from a reaction is time-consuming, and some material is inevitably lost during purification, so linking several steps in one pot can be a much more efficient way to make molecules. The saving can really add up if you can successfully design a three or even four step cascade.
’This is where I think organic synthesis will go,’ says Dieter Enders, who works on reaction cascades at RWTH Aachen, Germany. ’No protecting groups, in one pot, mild conditions, one catalyst controlling several asymmetric bond formations. We want to do it like Mother Nature.’
MacMillan says that his group has now done a (as yet unpublished) quadruple organocascade in the lab, and is working on a cascade of five. But it isn’t easy. ’Every time you add another catalytic cycle, I think it takes a year from your life!’ he laughs. ’But the pay-off is just enormous, because of the incredible complexity you can build, in a controlled fashion, from absolutely nothing.
’One of my goals for the next few years is to try to convince the community that this is a reasonable thing, and we should be doing it. Once you get into triple or quadruple cascade catalysis, it gets much trickier to figure out how it will all play out. The way to convince the community is with the pay-off. Once we make three, four, hopefully five natural products, I think then everyone will say that this is an exciting way to go.’
MacMillan has already started making natural products using this technique, making (-)-flustramine B via a double cascade, coupling two component molecules together in the first step and then cyclising them in the second.5 An elegant strategy, says Enders.
Still, in many areas of organocatalysis there is some disagreement about who exactly came up with and published a concept first. ’There’s a lot of work in the literature prior to David’s coining of the term ’organocascade’,’ says Barbas. ’But like ’organocatalysis’, I think that the name ’organocascade’ sort of draws everyone in to this concept. We’d called these reactions ’organocatalytic asymmetric assembly reactions’ - I guess that wasn’t catchy enough.’
Hot stuff
Naming isn’t the only area of controversy in the field. ’Organocatalysis is perhaps a little too hot!’ Barbas told Chemistry World. ’It’s very difficult to have a project of your own that you can really see to the end without several other groups jumping in.’
This competitive field has recently seen high-profile examples where the rules of fair play between scientists have been bent. ’There’s been a lot of difficulty with disclosing reactions before you publish them and people going back and doing the same reaction and trying to publish it before you,’ Barbas comments.
In March this year, Armando Córdova, an organocatalysis chemist at Stockholm University, Sweden, was sanctioned by the university for research misconduct. Commissioned by Stockholm University to investigate Córdova’s actions, organic chemistry professors Torbj?rn Frejd and Olov Sterner of Lund University found four separate cases of what they described as ’unethical behaviour’.
In particular, Frejd and Sterner found that Córdova, after hearing a talk by Donna Blackmond of Imperial College, London, took Blackmond’s ideas and published them as his own. The report adds that, while Córdova has subsequently demonstrated ’a confused and incorrect’ understanding of the underlying concepts, he ’continues to attempt to claim some credit’ for the work.
The report also highlights separate cases where Córdova ’has found it acceptable to hide or even omit references to relevant work of others, and we believe that it has been intentional.’ Córdova insists that any overlap of ideas or omissions of credit were not deliberate.
’The whole area has become so competitive because there are lots of people essentially doing the same thing,’ says Gaunt. But he doesn’t believe the problem is widespread. ’I think that the area has been tarnished and misrepresented by one or two examples. That’s unfortunate and it does have an impact on the rest of the field.’
Barbas suggests that the rush to get into the field illustrates the power of the chemistry itself. ’It’s so easy and simple to do because the catalysis works on unmodified substrates [unlike most metal-based catalysis]. That is what has drawn so many people into it,’ he says.
And List points out that organocatalysis allows synthetic organic chemists to make their own catalysts. ’Finally, those people that actually use catalysts are able to really understand and design and make [their] own.’
Perfect reactions
MacMillan isn’t content to sit back and admire the progress made so far in organocatalysis, which has tended towards improving known classes of reaction. ’We need to be constantly going out and trying to find new types of reactivities,’ he says. ’It’s a very challenging thing to come up with them, but so worth it - the minute you see it, the minute it works, you can see its going to open the door to many new reactions.’
Gaunt agrees. ’If you think of what the mainstay of organocatalysis has been so far, it’s been focussed on a number of reactivity principles that are actually well established,’ he says. ’An example of how this work can move forward to the next stage is MacMillan’s work on SOMO catalysis. That opens up a new ballpark.’
SOMO, so good
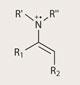
Published by MacMillan just this year, ’SOMO’ catalysis, which stands for singly occupied molecular orbital, combines organocatalysis with free radical chemistry.6
Free radicals are molecules (or atoms) with an unpaired electron, and tend to react quite differently to molecules with all their electrons in pairs. This different reactivity means a new series of molecular transformations becomes available. SOMO catalysis involves generating an enamine, and then stripping away a single electron to form the free radical intermediate, which will react with a whole new set of coupling partners.
Where enamine intermediates react with electron-poor molecules, SOMO intermediates react with a complementary set of electron-rich compounds.
’Ultimately,’ says List, ’what we try to accomplish is developing really perfect reactions, like for example Suzuki coupling, or olefin metathesis - dream reactions that work with almost any substrate combination that you try, and that always give high selectivity and high yield.’
There’s still some way to go before the sequence could be scaled up, says MacMillan. ’I don’t think it’s yet made a manufacturing process, but I expect that will come very soon,’ he says. ’Once you’ve done that, you’ve achieved the level of success that reactions like metathesis have achieved - and metathesis is just one reaction, this is a whole field.
’The fact that everyone is doing organocatalysis is great,’ he concludes. ’At first you start to get testy because you think people are moving into your territory. Then you recognise that it’s not about territories, it’s about the greater good of the community - it’s about as many people as possible getting involved.
’I find it incredibly satisfying, and exciting at the same time,’ he says. ’And I think the best stuff is still to come.’
Additional reporting by Mark Peplow
References
1 K A Ahrendt et al, J. Am. Chem. Soc., 2000, 122, 4243
2 B List, R A Lerner and C F Barbas, J. Am. Chem. Soc., 2000, 122, 2395
3 Z G Hajos and D R J Parrish, J. Org. Chem., 1974, 39, 1615
4 Y Huanget al, J. Am. Chem. Soc., 2005, 127, 15051
5 J F Austin et al, Proc. Natl. Acad. Sci. USA, 2004, 101, 5482
6 T D Beesonet al, Science, 2007, 316, 582











No comments yet