
Emma Stoye has the full story of how Frances Arnold, George Smith and Greg Winter put evolution to work in the lab
Earlier this year, Frances Arnold told us ‘nature is the best chemist of all time’. You only have to look at the diversity of life to see what she means. Over billions of years, evolution has given rise to an almost infinite variety of molecules, reactions and chemical pathways that help living things inhabit virtually every corner of the planet.
The three recipients of this year’s Nobel prize in chemistry have, as the Nobel committee put it, ‘harnessed the power of evolution’ at the molecular level. Arnold’s work on the directed evolution of enzymes has created biological systems with completely new chemistries and enabled the production of biocatalysts for greener chemical manufacturing. She receives 50% of the prize, with the remaining half shared between George Smith and Greg Winter for their pioneering work on phage display, a way of evolving new proteins and antibodies that has ultimately led to the development of drugs that have improved the lives of millions.
It is the ‘broad and profound impact’ that directed evolution strategies have had across various scientific disciplines that makes them worthy of a Nobel prize, says Rudi Fasan, a former post-doc of Arnold’s who now runs his own lab at the University of Rochester in the US. ‘They have revolutionised the way we think,’ he tells Chemistry World.
Age of the phage
Back in the 1980s, George Smith had no idea his work would eventually lead to advances in medicine.

‘My vision at the time was pretty narrow,’ he said at a post-Nobel prize announcement press conference at the University of Missouri, US, where he is an emeritus professor. ‘I was specifically looking for a much easier way to search through big libraries of proteins that would bind an antibody.’ He was keen to stress that his discovery was heavily dependent on lines of work that had gone before. ‘I happened to be in the right place at the right time.’
That place was in the lab of biochemist Robert Webster at Duke University in the US state of North Carolina, where he spent two years working on sabbatical from Missouri. At the time, researchers had succeeded in building vast libraries of random peptides and proteins, but searching through these for a protein with a desired activity – binding to a particular antibody or target, for example – was slow and laborious, as each molecule would have to be screened individually. It was also difficult to link particular proteins with the genetic sequences that encoded them.
I happened to be in the right place at the right time
George Smith
Smith worked out a way to overcome these hurdles using filamentous phages – bacteria-infecting viruses whose simple physiology had already been well studied. Phages consist of genetic material surrounded by a protein coat – the phage inserts its own genes into the bacteria’s DNA so that it is replicated to produce more phages.
He showed that if a gene fragment was inserted into the phage’s DNA in the middle of a gene for one of its own coat proteins, it would be copied during replication and the peptide or protein it coded for would end up being ‘displayed’ on the surface of the phage, hence the name of the technique. He reasoned that you could then search through a mixture of phages and ‘fish out’ those carrying a protein that binds strongly to a particular target. Because the phage would contain the original genetic sequence, this was also a way to identify an unknown gene for a protein of interest.
In 1985 Smith demonstrated this for the first time using a 57-amino acid long fragment of a restriction endonuclease inserted into the filamentous phage genome within gene III, which codes for one of the coat proteins. He showed an antibody that bound to the peptide fragment could be used to capture the phages containing the foreign peptide out of a pool of others.1 In the following years he refined the method and showed that repeated rounds of phage display could be used to select proteins with the highest binding affinity for an antibody target.2
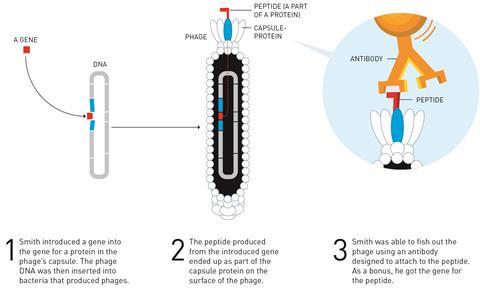
‘Phage display would allow you to create huge libraries of clones with ten billion or a hundred billion structures in it, and instead of having to search through them months at a time you develop a way of selecting, physically pulling out the ones you want so you can throw away the others,’ Smith said at the press conference.
Paul Dalby, a professor of biochemical engineering at University College London in the UK remembers using methods developed by Smith while working on a phage display project as a post-doctoral researcher at the University of Philadelphia in the late 1990s.
‘The first thing [I was given] was a copy of George Smith’s protocol notes – it was a photocopy of a photocopy of a photocopy that had come out of his lab,’ he remembers. ‘It’s a bit like a bible for phage display – everybody started using that technique.’
The technology was rapidly adopted by other groups studying proteins, who began to take it in new directions. Meanwhile, Smith’s own work moved away from phage display in the 1990s around a decade after he first demonstrated it. At that time he still had no idea of the impact his contribution would eventually have.
‘Smith overcame a lot of technical barriers to get that system to work,’ says Dalby. ‘But it was just a technology for displaying proteins and selecting out peptides originally.’
It was Winter’s work at the Laboratory of Molecular Biology in Cambridge, UK, that would enable phage display to kick-start a revolution in drug development.
Nature’s defences
‘I didn’t know how much to believe in antibodies – to the pharmaceutical industry, medicines were chemicals, small molecules,’ says Winter. ‘But I thought it’s got to be a good idea – they are our normal route for protecting us against viruses, bacteria and pathogens, so why can’t we turn them against cancer, for example?’

Smith’s earlier work had focused on manipulating the structures of enzymes. But his interests shifted to antibodies under the influence of Georges Köhler and César Milstein at the LMB, who received the 1984 medicine Nobel for developing monoclonal antibodies – those made by genetically identical immune cells.
He knew that all antibodies shared the same basic structure, with relatively small differences responsible for sites that bind to different targets. For several years his work focused on using this ‘scaffold’ to engineer new proteins. It was this knowledge of the structure of antibodies that eventually allowed him to apply the principles of phage display. At the time, no one had displayed anything as complex as an antibody – which comprises more than one polypeptide chain – on the surface of a phage, so Winter used the sequence for a fragment of the antibody which contained its binding region. This meant that the binding site of the antibody would be displayed on the surface of the phage when the sequence was inserted into the gene for one of its coat proteins, and could be fished out using the binding target – a molecule called phOx – in a sort of reversal of Smith’s original experiments: part of the antibody is on the phage and the selection is done by the target. Winter showed this could work in 1990, achieving the first ever phage display of a folded and fully functional antibody fragment.3
Just as with proteins, this paved the way for creating huge libraries of antibodies, and Winter went on to show that phage display could be used to evolve antibodies with a much higher binding affinity for the target than those obtained by traditional immunisation methods, using repeated rounds of mutation and selection to develop high-affinity antibodies that retained their specificity for the target.4
Together with colleagues he formed a spin-out company, Cambridge Antibody Technology (CAT), to focus on antibody therapeutics. After undergoing development throughout the 1990s, the world’s first drug based on a human monoclonal antibody – Humira (adalimumab) – was approved in 2002.
New therapies
Humira binds to and inhibits tumour necrosis factor alpha (TNF-α), a cell-signalling protein that promotes the inflammation associated with auto-immune diseases. It was originally developed to treat rheumatoid arthritis but has since been approved for use against a number of autoimmune conditions including Crohn’s disease, psoriasis and ankylosing spondylitis, becoming a mega-blockbuster that is still the world’s top-selling drug, achieving revenues of nearly $18.5 billion (£14 billion) in 2017.
The advantage of antibodies is they’ve got a structure that makes them very amenable to change
Greg Winter
‘The development of Humira transformed a lab-based technique into one which was accepted for drug development,’ says Jane Osbourn, who has been at CAT since the early 1990s and is now vice-president of research and development at its successor MedImmune, which is owned by AstraZeneca. ‘[It] really sealed the reputation of antibodies as therapeutics within a community that was previously sceptical.’ She says Winter was ‘an enormous presence in CAT’s early days – a brilliant, science-focused and inquisitive leader. He was always tracking the latest developments and inspiring us all to do more.’
Today, phage display is widely used in pharma and biotech, and there are many antibody therapies available to patients or in clinical trials. ‘The technology can be applied to any disease, from oncology to asthma to diabetes, and so on. It has helped us to understand that we can potentially make an antibody to target any protein,’ says Osbourn.
In future, researchers will continue to generate ever-more exotic antibody fragments with additional functions, says Winter. ‘The advantage of antibodies is they’ve got a structure that makes them very amenable to change. They’ve got lots of domains so it’s easy to plug another one on.’
In particular, he says, the emergence of bi-specific antibodies, those capable of binding two therapeutic targets is exciting. ‘Bi-specifics can bring two types of cells together, so you could bring T-cells to any particular target, and we’re seeing those already. One very interesting use of that recently is [Roche’s] bi-specific Hemlibra, which mimics Factor VIII in people with blood clotting diseases. I think there are a number of situations where we can create antibodies that mimic a natural protein.’
Evolving enzymes
While the invention of phage display allowed researchers to produce new proteins and antibodies, it was Arnold’s work in the 1990s that showed you could evolve an enzyme.

‘Up to then you could use phage display to make variants of things that bound an antibody or target, but what we couldn’t do was take that same technology and apply it to enzymes so that they catalyse a reaction better,’ says Dalby, who spent time in the 1990s trying to adapt phage display to enzymes. This is because, he explains, enzyme activity is determined by characteristics it is impossible to fish out using phage display, such as how many substrates it converts in a second. ‘Frances Arnold came up with a parallel technique of using the same concept. She brought this new field out, and it took off really rapidly.’
Arnold did not begin her career as a molecular biologist. After graduating as a mechanical and aerospace engineer she worked on designing solar energy facilities, before turning her attention to proteins. At first she was interested in rational design – trying to directly rebuild proteins’ structures to make them function differently. She describes being ‘blissfully ignorant’ of the challenges this would involve.
‘I didn’t realise how difficult it would be to engineer better proteins. This was the beginning of the DNA revolution and we were just learning how to manipulate DNA and the code of life for human purposes, but we didn’t understand the rules,’ Arnold tells Chemistry World. She began to consider evolutionary approaches – which had been theorised by German biochemist Manfred Eigen in the 1980s but never successfully demonstrated – when ‘desperation set in’, she says.
‘It just seemed apparent to me that you could circumvent ignorance of how sequence encodes function by making mutations at random and letting the system show you what the rules are,’ says Arnold. ‘The timing was right because the methodology was there – Jerry Joyce had published error-prone PCR methodology which was very useful for mutating [and] new screening capabilities had become possible, so I could assay hundreds of enzymes in the time it would usually take to assay one.’
We found beneficial mutations that would improve the enzyme activity that were 20–30Å away from the active site
Frances Arnold
In the first demonstration of directed evolution of an enzyme, Arnold developed a version of the enzyme subtilisin E that was able to work in a very different environment to the one it was adapted to – in the presence of an organic solvent. She used a version of the polymerase chain reaction (PCR) to introduce random mutations into the genetic sequence for the enzyme and tested thousands of variants for their ability to break down the protein casein in a 35% solution of dimethylformamide, selecting the most effective and subjecting them to further rounds of mutation and selection. Within three generations she identified a version of subtilisin with 10 mutations that worked 250 times better in DMF than the original.5
Arnold knew almost immediately she was onto something special. ‘I knew it would be important [because] the mutations that conferred benefits were surprising. All the so-called rules that people had come up with for how you might engineer a protein could not account for how the protein thought it should be engineered,’ she say. ‘We found beneficial mutations that would improve the enzyme activity that were 20–30Å away from the active site, for example, or stabilising mutations that were on the surface of the protein. That seems obvious now but it wasn’t then.’
A new kind of catalysis
Arnold went on to show that various properties of enzymes – including the temperature at which they function best, their stability or their catalytic activity towards a particular substrate – can be changed using a speeded up version of the process that happens during their natural evolution: creating variation in a ‘parent’ enzyme by introducing genetic mutation, selecting the most effective ‘offspring’ through screening, and repeating the process.
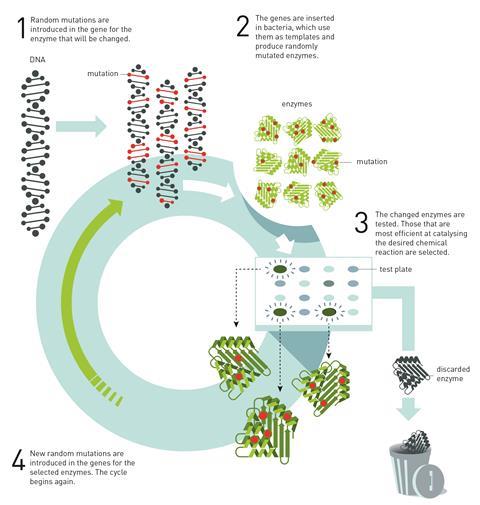
Fasan says Arnold’s initial methodology was technically straightforward to implement, which meant other researchers were quick to adopt directed evolution to alter a variety of enzymes. ‘Since then, these techniques have been further developed in many ways and for addressing other types of problems, such as adapting an enzyme to recognise a non-native substrate and/or improving its chemo-, regio-, and/or stereoselectivity in the context of a particular chemical transformation,’ he says. One area rapidly growing in importance, he says, is the use of evolved enzymes for drug synthesis. Laboratory-evolved enzymes have so far been used in place of industrial catalysts to produce intermediates during the manufacture of the cholesterol-lowering drug Lipitor (atorvastatin) and the antidiabetic drug Januvia (sitagliptin), for example.
‘The sitagliptin case is particularly impressive,’ says Fasan. ‘A biocatalytic process involving a laboratory-evolved transaminase enzyme can replace a well-established chemocatalytic process for the preparation of a key intermediate of this drug, bringing about a significant increase in productivity while reducing the waste associated with the entire process, as well as eliminating the use of toxic metals.’
Green machines
An area that particularly excites Arnold herself is the use of evolved enzymes for green chemistry. ‘I want to make enzymes that can do useful chemistry that currently is being done in a non-sustainable fashion,’ she says. ‘My ideas come from human-invented chemistry. I look at what people have done with iridium or rhodium and I say “Could a biological system do that if I trained it well?” And the answer is yes – a lot of this chemistry can be encoded in DNA.’
Returning to her original ambitions to work on renewable energy, Arnold has developed biocatalysts that can enable microbes to convert simple sugars to potential biofuels such as isobutanol, directly evolving two enzymes within a biosynthetic pathway to use a by-product of the normal metabolism of E. coli as a cofactor.6
This chemistry is possible, and maybe the enzymes are the most efficient way to do this
Frances Arnold
In a more recent development that made headlines around the world, her group recently succeeded in making the first enzyme that can catalyse the formation of carbon–silicon bonds, a reaction not seen in nature. To do this, they evolved a cytochrome c from the hot spring bacterium Rhodothermus marinus, which was capable of catalysing carbon–silicon bond-forming reactions but not as its primary function, and with extremely low efficiency. After several rounds of targeted mutagenesis and selection the enzyme could catalyse the insertion of carbenes into silicon–hydrogen bonds 40 times better than the starting enzyme and, excitingly, at much higher efficiency than the best known synthetic catalyst for the same reaction.7
By fine-tuning enzymes to catalyse new reactions, Arnold hopes we will eventually see a future where the traditional chemical factory has been replaced by biocatalytic microbes able to carry out industrial processes in a sustainable way, free from heavy metals.
‘My group has shown over the last few years that simple, Earth-abundant iron [in enzymes] can catalyse reactions that people only previously catalysed with iridium, for example. And that’s wonderful for all of us, because it shows us that this chemistry is possible, and maybe even the enzymes are the most efficient way to do this.’
Rational meets random
Directed evolution is by no means limited to enzymes that exist in nature. Researchers are starting to show it can be combined with computational approaches that are used to engineer completely new proteins and enzymes from scratch.
‘Computation has come a long way, particularly for the design of proteins, but function is still hard, and that’s where evolution really helps because it can take us over the finish line,’ says Donald Hilvert of the Swiss Federal Institute of Technology in Zurich, whose group has collaborated with computational protein designers, such as David Baker at the University of Washington in Seattle, US, to create biocatalysts that carry out useful reactions not found in nature, including aldol condensations and 4+2 cycloadditions.8
‘The computation provides us a foot in the door but the activities are much lower than you would associate with a very active natural enzyme,’ Hilvert says. ‘We’ve applied iterative cycles of mutagenesis and screening to optimise their properties. In some cases we can improve the starting activities by five orders of magnitude. Often what we find in the course of evolution is that the original design can be overthrown in favour of something that’s more effective, so evolution can be quite creative in telling us how to fulfil a particular task.’
I’d like to do my work, but I also want to support science done by younger people
Frances Arnold
The field has been further boosted by high-throughput screening assays that have sped up the selection process, as well as advances in how the enzymes are mutated in the first place, he says. ‘You can do things completely randomly or you can focus your attention at particular regions of a protein. You can randomise the active site or specific stretches of an amino acids in a protein.’
Further technological advances will bring about yet more improvements to the way enzymes can be designed, Hilvert believes. ‘There are enormous opportunities for designing new materials and new catalysts. I think we’re just at the start.’
Arnold agrees we’re not even close to the limits of what directed evolution can achieve. After winning the Nobel prize, she is keen to carry on leading her research group, though she also hopes the attention from the prize will inspire up-and-coming researchers to adopt some of her lab’s techniques. ‘I’d like to do my work, but I also want to support science done by younger people and I can do that in a positive way now,’ she says. ‘There are a lot of young chemistry professors who think this is a really interesting way to go, and their creativity will result in some very interesting transformations.’
References
1 G P Smith, Science, 1985, 228, 1315 (DOI: 10.1126/science.4001944)
2 J K Scott and G P Smith, Science, 1990, 249, 386 (DOI: 10.1126/science.1696028)
3 J McCafferty et al, Nature, 1990, 348, 552 (DOI: 10.1038/348552a0)
4 T Clackson et al, Nature, 1991, 352, 624 (DOI: 10.1038/352624a0)
5 K Chen and F Arnold, Proc. Natl Acad. Sci. USA, 1993, 90, 5618 (DOI: 10.1073/pnas.90.12.5618)
6 S Bastian et al, Metab. Eng., 2011, 13, 345 (DOI: 10.1016/j.ymben.2011.02.004)
7 S B J Kan et al, Science, 2016, 354, 1048 (DOI: 10.1126/science.aah6219)
8 R Obexer et al, Nat. Chem., 2017, 9, 50 (DOI: 10.1038/nchem.2596)









1 Reader's comment