
Katrina Krämer tells the story of how click and bioorthogonal chemistry came to win the 2022 Nobel prize
Discovering reactions is synthetic chemists’ bread and butter. The best reactions are simple to do, fast and robust, high-yielding, wide in scope and free of by-products. ‘That is obvious and does not constitute an idea – it is merely common sense,’ an anonymous organic chemists told Chemistry World for an article back in 2007.
But this common sense has now earned Carolyn Bertozzi, Morten Meldal and Barry Sharpless the 2022 chemistry Nobel prize for the development of click chemistry and bioorthogonal chemistry. What made their achievement so special is the ability to see through the complexities of making molecules and prune down chemical concepts to what’s really important. The reactions they discovered are so simple yet effective that they can be carried out even in the chemical chaos that exists inside living systems.
Click chemistry might look effortless but ‘it’s not simple chemistry, it’s incredibly sophisticated to discover’, says M G Finn, who helped develop the concept as part of Sharpless’s team at the Scripps Research Institute in California, US. The path to this year’s prize required the laureates to connect the dots between a chance discovery and half-forgotten ideas from chemical history to create reactions that can be run in cells, animals and even humans, just as well as they can be run in a flask.
In 2001, Sharpless, Finn and Hartmuth Kolb laid out why synthesis needed a new, simplified concept in an 18-page essay. Chemists, Sharpless argued this year, spend too much time trying to recreate the devilishly complex compounds produced by organisms with a billion years’ evolutionary head start. ‘Nature is a matchless creator of carbon–carbon linkages and we propose leaving the tough job of carbon–carbon bond synthesis as much as possible to her,’ the team wrote.
Instead of trying to directly copy natural structures and reactions – and often doing so badly – researchers should consider nature’s approach to creating molecular complexity: a set of simple yet powerful, highly reliable and selective reactions. The question was, says Finn, ‘Can we develop chemical reactions that people can do with the same reliability that biology has after billions of years of evolution and stitching together small building blocks?’
Why are you giving a new name to just good chemistry?
By focusing on just a few, nearly perfect ‘spring-loaded’ reactions, chemists could access structures with any function they wanted – even if those structures aren’t exact copies of natural products. As long as the compound has the molecular properties one is after, its exact structure becomes essentially irrelevant. ‘Really, what matters is function,’ says Finn, who at the time was Sharpless’ faculty colleague after doing a PhD in his group. ‘What are you making, what does it do? It takes the attention away from how you make the bonds to why you are making them. That’s all that biology does, it’s a relentless function-driven machine. As people in the laboratory trying to invent new things, that’s a strategy that we could well adopt.’
The team gave this philosophy the name click chemistry, meant to capture the satisfaction and ease of snapping together a push buckle or clicking in place a perfectly fitting puzzle piece. But people asked ‘Why are you giving a new name to just good chemistry?’ recalls Finn. In some sense, this was fair criticism, he says, but it missed an important point. ‘Driving this concept to its ultimate would … really enable non-chemists to make new molecules in a way that was well aligned with their goals and with the resources they had available.’ People wouldn’t have to rethink their procedure during scale-up or when adding a new functional group into their molecule.
The original clicks
To classify as click, a reaction has to adhere to a set of extensive and stringent criteria including being insensitive to oxygen and water, taking place in no or benign solvents, being stereospecific and selective for a single product that requires no chromatography to isolate, and having a thermodynamic driving force greater than 20kcal/mol. The trio identified a few reactions that would satisfy these criteria. Among them are urea and amide formations, nucleophilic ring openings of strained heterocycles such as epoxides and aziridines, and the transformation that would become almost synonymous with the click concept: the 1,3-dipolar cycloaddition between azides and alkynes to make triazoles.

The reaction has a long history, with the first examples dating back as far as 1893 when Arthur Michael – discoverer of the conjugate additions that bears his name – reacted phenyl azide with the alkyne dimethylbutyndioate to make 1,2,3-triazole. In the early 1960s, Rolf Huisgen took a closer look at reactions between 1,3-dipoles and dipolarophiles to form five-membered rings, which is why this reaction is now often called Huisgen cycloaddition.
In their 2001 essay, Sharpless and co-workers described the Huisgen cycloaddition between an azide and acetylene as ‘about as good as a reaction can get’. Azides, they argued, have the spring-loaded nature required for click chemistry yet they remain inert towards pretty much all other functional groups other than dipolarophiles. Moreover, azides are stable under most conditions including in water and air. And the resulting triazole is essentially inert to oxidation, reduction and hydrolysis.
But Huisgen cycloadditions had fallen out of favour over the years. Maybe it was because of safety concerns: azides have to be handled with care, and under certain conditions reactions can produce extremely explosive compounds like hydrazoic acid or diazidomethane. Maybe it was because they could produce regioisomer mixtures, or because most required temperatures well above ambient and were very slow – even Sharpless’ first attempts required up to 130°C for at least 40 hours. ‘It’s probably one of the least reactive dipolar cycloadditions,’ says Valery Fokin from the University of Southern California, US.
Fokin, who had joined the Scripps faculty in 2000 after a postdoctoral stint in Sharpless’ lab, had an interest in metal acetylide chemistry, which stemmed from his PhD work. ‘I was looking at, specifically, the stability of the copper acetylides in aqueous solutions – what they are, how they behave, how we can capture them with electrophiles. Azide was a perfect electrophile for that,’ he explains. Sloppier syntheses, Fokin recalls, including wet solvents and no inert atmosphere, surprisingly often resulted in better outcomes. One day in late 2001, he set up reactions between phenylacetylene and an azide in an alcohol/water mixture, adding different metal catalysts – iron, zinc, nickel, silver and copper. At 7pm, a few hours after starting the experiments, he wrote excitedly in his lab notebook ‘The copper reaction looks good by TLC! LCMS is OK, definitely some triazole there! Very cool!’
‘Within a few days, I was [running the reaction] in my own plasma and in my own blood, just a little bit of copper sulfate and ascorbate, which I got from [supermarket] Trader Joe’s,’ recalls Fokin. While he had first used copper(i), he quickly settled on reducing a cheaper copper(ii) source in situ after thinking back to a high school experiment in which copper(ii) is reduced with ascorbate (vitamin C). But Fokin would later find that even solid copper wire could catalyse the reaction.

‘It was just spectacular how well it worked with some of the substrates – the isolation, the purity and just how little catalyst was required to get it to work in high yield, high regioselectivity,’ says Seva Rostovtsev, who worked as a postdoc on the project together with Sharpless, Fokin and Luke Green. The reaction produced almost quantitative yields of a single triazole isomer, no matter what functional groups were attached to either reaction partner. It required little more than throwing the ingredients together, stirring for a while and filtering off the product. ‘No reaction is absolutely 100% efficient, there are always limitations,’ Fokin points out. ‘Just in this case the limitations were surprisingly few.’
‘The CuAAC [copper(i)-catalysed azide–alkyne cycloaddition] reaction cannot be stopped,’ Sharpless said in a press conference after the Nobel announcement. ‘It’ll go 100% every time. You can have it in urine. You can have it in minestrone soup.’ Apart from strong catalyst poisons – thiols, sulfides, hydrogen cyanide – there’s very little that can shut down the reaction. ‘The beautiful thing about the reaction is that it really is hard to screw up,’ says Finn.
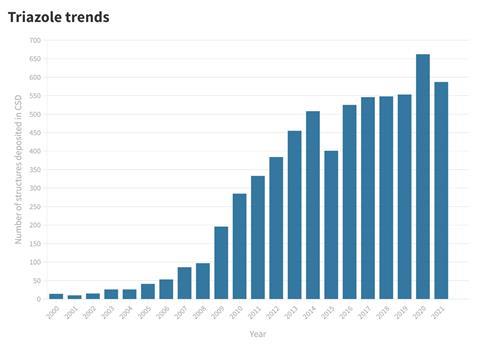
So simple and efficient was this transformation that the team was surprised nobody had discovered it before. ‘The fact that this “unstoppable” reactivity of copper acetylides with organic azides remained unrevealed until now, despite the great body of research on copper-mediated organic synthesis over the last 70 years, is extraordinary,’ the researchers wrote when they first described the reaction. Maybe the reaction was just waiting to be discovered, as on the other side of the Atlantic another team ran into it at almost exactly the same time.
Clicking independently
Morten Meldal was working with PhD researcher Christian Tornøe at the University of Copenhagen in Denmark trying to develop organic reactions that could be run on solid phase akin to peptide or DNA synthesis. ‘One of the reactions that we were trying out was the acylation of alkynes, copper(i)-catalysed,’ Meldal told Chemistry World. ‘I was going to bring alkynes together with acid chlorides,’ explains Tornøe, who is now a principal scientist at the pharma company Novo Nordisk in Denmark. ‘By chance, I had an azide hanging off the acid chloride.’ The azide was, in fact, a way to protect an amine that would otherwise interfere with the reaction.

But things didn’t go as planned. ‘When Christian did that reaction, he got a by-product, he never could get his acyl chloride to couple,’ Meldal recalls. ‘He came down to me and was initially really devastated because his project didn’t work.’ But instead of throwing it out, the team took a closer look. ‘We realised that this was very, very fortunate what we had discovered,’ Tornøe says. The reaction was amazingly clean and efficient. ‘We called it QCT reaction – quantitative chemical transformation,’ Meldal says. ‘This never caught on because click is of course much nicer.’
It took the team only a few months to turn their serendipitous discovery into a workable methodology, compatible with solid-phase peptide synthesis. ‘We immediately started to utilise this in an orthogonal fashion to make chemistries that were probably impossible in all other ways,’ Meldal recalls. ‘When we presented it as a poster at the American Peptide Symposium, there was just standard interest from people at the conference. But maybe peptide chemists are not that interested in alternative chemistries,’ he laughs.
Meldal’s and Sharpless’s papers came out within a few months of each other. At the time, neither team knew of the other’s work, though their reaction would soon make waves in the synthesis community. ‘I remember running the copper reaction for the first time in my life,’ says Jiajia Dong from the Chinese Academy of Sciences in Shanghai, who would later join Sharpless’s team. ‘I saw the result coming out of the LC-MS. “Holy cow,” I said, “it’s only one peak – I never saw any reaction like this!”’ The stark difference between click chemistry and many other transformations is obvious to every chemist who ever had to do reaction optimisation. ‘In 2019, we did reactions with 1200 azides under the same conditions – and almost every one of them was quantitative in yield,’ Dong says.

It quickly became clear that the copper-catalysed cycloaddition would not be confined to the hands of synthetic chemists. ‘We became immediately aware that the copper reaction was going to be something special when material scientists were the first ones to really jump on the bandwagon,’ says Finn. ‘Material scientists, all they care about is making useful stuff. They’re not so concerned with the elegance of the chemistry. When they started using this right away, that was a signal.’
Finn’s team was among the first to use the reaction in a biological system, to functionalise virus-like particles. ‘There was no chemistry that anybody knew about that could actually do that at the time – we tried a lot of different reactions,’ Finn says.
‘I’m amazed by the creativity in the field,’ Tornøe says. ‘[Click chemistry] has been applied to stuff that I would never envision, much more broadly outside organic chemistry.’ ‘Today, there’s no chemist with respect for themselves who doesn’t do click chemistry once in a while,’ says Meldal.
Biology beckons
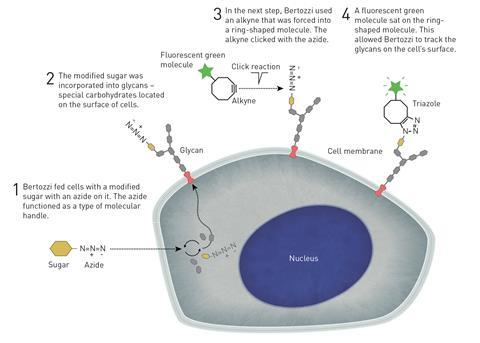
The reaction quickly made the jump from glassware to cells. ‘In about 2002, we published the synthesis of a small molecule, and we really wanted to know a little bit more about how it worked, look at where it went inside cells,’ recalls Alison Hulme from the University of Edinburgh, UK. ‘Click chemistry was just coming into fruition, and now we could add this tiny little handle to our small molecule, it will still be perhaps active inside cells, but then we could click on a fluorophore so we could visualise it.’ However, since copper ions to are toxic to cells, the classic azide–alkyne reaction would remain of limited use in living organisms. It would take Carolyn Bertozzi and her team at the University of California, Berkeley, US, for click chemistry to become truly bioorthogonal – running under physiological conditions without interfering with, or being affected by, surrounding biological processes.

At around the same time as Sharpless and his team were laying out the idea of click chemistry to a still sceptical chemistry community, Bertozzi was trying to solve what first looked to be an entirely different problem: how to study cell surface sugars. Glycans and glycoproteins play an important role in immune responses. But while there were powerful imaging tools for proteins and nucleic acids including in living cells and animals, looking at glycans in their natural cellular environment wasn’t possible – the cells had to be destroyed to profile the sugars. What was really needed was a reaction that wouldn’t interfere with cellular processes but would still reliably and selectively target glycans.
It’s hard to overemphasise how complex cells are compared with the clean environment of a reaction flask. ‘I’ve got a picture of me at the Taj Mahal, it’s me with the building in the background – and then I’ve got a picture of what it was actually like, the big queue of people in front of me, masses of people,’ says Hulme. ‘Most people’s image of doing a reaction inside a cell is like the first picture: there’s your target, there’s your substrate and they just get there. But the reality is like the second picture: it’s this crowded environment. It’s really complicated. There are things going on all the time.’
But Bertozzi and PhD researcher Eliana Saxon managed to repurpose an old reaction to cut through the chemical chaos. Their reaction of choice was the Staudinger ligation, a reaction between an organic azide and a phosphine was discovered in the early 20th century. The classic reaction produces an iminophosphorane or aza-ylide, which is highly unstable in water and hydrolyses almost immediately. But Bertozzi and Saxon designed a phosphine with an electrophilic trap that, instead of falling apart in water, does an intramolecular cyclisation and leaves behind a stable amide bond.
In biological systems, you’re often dealing with components present at very low concentrations – you really do need fast reactions
Now all the team needed to do was to get azide-modified sugars into cells, which was as easy as feeding cells synthetic azidosugars. Nature doesn’t seem to use or produce azides, so the inconspicuous little functional group is essentially ignored by cells. ‘Enzymes are evolved to recognise things that they’re used to seeing,’ Hulme explains. Once cells had taken up the azidosugars and incorporated them in their surface structures, the team could add their biotin-linked phosphine. Only a few years later, the team proved that they could do the reaction in living mice.
‘I don’t think that anyone appreciates how clean and how amazing that reaction is until you do it on a protein lysate or in an animal, it just works amazingly well,’ says Ellen Sletten, now at University of California, Los Angeles, who did her PhD with Bertozzi at Berkeley. ‘It was really the first bioorthogonal reaction.’
But while the Staudinger ligation remains one of the most selective bioorthogonal reactions, it remained too slow for many dynamic processes. ‘In biological systems, you’re often dealing with components that are present at very low concentrations: micromolar, maybe nanomolar concentrations,’ explains Nicholas Agard, who did his PhD with Bertozzi and now works for US biotech company Genentech. ‘In order to capture a lot of what’s present at that those very low concentrations, you really do need fast reactions.’
The more the team tried to speed up the Staudinger ligation by creating more nucleophilic phosphines the more prone they became to non-specific oxidation, recalls Jennifer Prescher of University of California, Irvine, who was a PhD researcher in Bertozzi’s lab at the time, working first on the Staudinger ligation in mice and then on strain-promoted click chemistry. ‘It just wasn’t going to be feasible.’
Meldal and Sharpless’s click chemistry would have been perfect – Bertozzi’s team was already using azides as one coupling partner in the Staudinger ligation so they would only have to swap the phosphine for an alkyne. But with copper’s toxicity, this didn’t seem like an option.
The click feels the strain
In the summer of 2003, Bertozzi had just come back from a physical organic chemistry conference, where people had been talking about ring strain. ‘The idea she had was: Could we use ring strain in some way to catalyse an azide–alkyne reaction?’ recalls Agard. ‘I thought this was fascinating.’
Agard dug into the literature and found one of Georg Wittig’s papers from 1961. ‘The paper was in German, and I speak no German. But the pictures looked good,’ laughs Agard. ‘I could read this one sentence: it said cyclooctyne, it said azide and it said explosion.’ Undeterred, Agard decided to make the compounds that Wittig had described as explosively efficient dipolarophiles in a cyclodaddition with azides.
The reason cyclooctyne and other cyclic alkynes are so reactive is that they carry a large amount of strain energy. After all, they are linear constructs trying to fit into a non-linear structure. Cyclooctyne is the smallest cycloalkyne stable enough to be isolated and stored – though it has an intensely unpleasant smell. ‘We always knew when they were being made because they smelled so bad, like rotting tissue,’ recalls Prescher.
By early 2004, Agard had succeeded in making a biotin-substituted cyclooctyne and Prescher was testing whether they would do the azide–alkyne click reaction in cells. ‘Seeing the shift on the histogram for the flow cytometry experiment indicating that the cells that were treated with the azidosugar and the cyclooctyne probe had a higher mean fluorescence intensity value – I will never forget that moment,’ Prescher recalls.
Within a few weeks, the team had published their discovery, though at the time they didn’t realise the impact it would make on bioorthogonal chemistry. ‘It was exciting to us as chemists at the time,’ says Prescher. ‘But did we think that this is going to blow up in the field? No, not at the time.’
Although the reaction initially wasn’t any faster than the Staudinger ligation, Prescher says the team saw more room for improvement in strain-promoted click chemistry. ‘People have increased that 1000-fold, 10,000-fold,’ says Agard.
Nobel networks
Science is seldom done by isolated individuals, and the work behind this year’s Nobel prize is no exception. We’ve taken each laureate’s publication history from Web of Science (going back as far as 1993) and plotted a network of people with whom they have published papers (limiting the search to original research papers and reviews only). For clarity, the laureates themselves are not shown in the visualisations.
Valery Fokin is a large node in Barry Sharpless’ network, shown in a paler colour indicating he has been collaborating with Sharpless since the late 1990s. And the collaboration is still going strong; Fokin is linked to many people who only began collaborating with Sharpless more recently (nodes in a darker colour). Hartmuth Kolb and M G Finn, who were co-authors on the 2001 essay setting out the concept of ‘click chemistry’, are also larger and more established connections. Sharpless also has a separate group of collaborators who are not connected to the others (to the far left of the visualisation). This is centred on fellow Nobel laureate Satoshi Ōmura of Kitasato University in Tokyo, Japan, with whom Sharpless has collaborated to investigate antibacterial compounds. Peng Wu is also a relatively large node who first published with Sharpless in 2003–2007 – he did his PhD with Sharpless, then a post-doc with Carolyn Bertozzi and is currently works at Scripps integrating synthetic chemistry with glycobiology.
Meldal’s network is notably less complex than Sharpless, perhaps reflective of generally smaller group sizes (and less funding) in Europe than in the US. Christian Tornøe, with whom Meldal developed their version of the click reaction, is a mid-sized node in the 1998–2002 bracket. He is connected to Frederik Diness’s larger node, one of Meldal’s colleagues in the University of Copenhagen chemistry department. The largest node is Klaus Bock, who was Meldal’s PhD supervisor at the Technical University of Denmark. When Bock later became the head of chemistry at the Carlsberg Laboratory, he persuaded Meldal to establish a research group – Meldal’s work on peptide synthesis here eventually led to discovering the CuAAC reaction.
As with Sharpless, Meldal’s network contains more pale nodes, indicating the well-established networks and long-standing collaborations of a chemist later in their career.
Bertozzi’s full network is incredibly complex, with over 1300 co-authors. The network shows several clusters of small nodes – these each represent one large multi-author, multi-institution collaboration that are typical of the more biological and medicinal research fields where Bertozzi’s work lies. In order to better see the details, we have also produced a visualisation showing only those collaborators who have appeared as co-authors with Bertozzi on at least two papers.
Even with those co-authors removed, Bertozzi still has a large and complex network. Julie Leary, who uses mass spectrometry to look at glycobiology at the University of California, Davis, is the largest node. Mass spectrometry is a key part of Bertozzi’s lab: postdoc Nick Riley is another mass spectrometry expert and prolific collaborator, producing over 20 papers with Bertozzi since 1999.
Compared to Sharpless and Meldal, Bertozzi’s network is also much newer and appears to be more interconnected.
Text by Neil Withers and Phil Robinson, data taken from Web of Science and processed by the RSC’s data science team (Colin Batchelor, Solenne De Pellegars and Anna Linfoot)
Clicking ahead
‘When [click chemistry] was first invented, it was still really organic chemistry: you needed a chemistry background in order to do [the reaction]’ says Caitlyn Miller, who works on cancer immunotherapy with Bertozzi, who moved to Stanford University in 2015. ‘Now it’s become so popular that a lot of commercial vendors are selling the linkers with the handles already available, and they’re even providing them as kits with protocols that biologists can follow. It’s been really cool seeing people being able to do this chemistry, and doing more cool biology with it than they would have been able to before.’ Watching click chemistry develop from research method to therapeutic application in people ‘has been a complete thrill’, says Prescher.
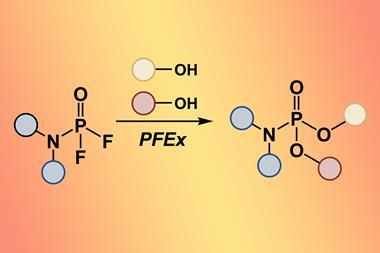
None of the three groups are resting on their laurels: Meldal’s team recently stabilised peptides with click triazoles in place of disulfide bonds. The Bertozzi group has found a new cellular component – glycoRNA – that nobody knew existed, and extended click chemistry into other hard-to-study cell components like lipids. And Sharpless’ team launched what they call the next generation click chemistry with the sulfur fluoride exchange reaction in 2014.
And there may be more click reaction hiding right under people’s noses. Meldal believes that ‘there’s a lot of reactions that by small modifications can actually fulfil the criteria of a click reaction’. But for the next big breakthrough, chemists will have to continue to challenge established thinking. ‘Chemistry, there’s so many things you can’t anticipate,’ Sharpless said in the press conference. ‘If you cover a lot of ground, you got a much better chance in the short life to hit some really good ground, where things aren’t at all the way we were taught.’
‘I think the diversity of people [I worked with during my early years at Berkeley] created an environment where we felt we didn’t have to play by the same old rules as scientists,’ Bertozzi said in a speech after the Nobel announcement. ‘We could do things like organic chemistry in living animals. Why not?’
Katrina Krämer is senior science correspondent at Chemistry World










No comments yet