
Efforts to cure malaria have been going on for hundreds of years. Clare Sansom looks at some of the latest – and most innovative
In 2015, the Nobel committee for physiology or medicine made the unexpected decision to award half the prize to Youyou Tu from the China Academy of Traditional Chinese Medicine in Beijing. Tu discovered an antimalarial drug that was later described as ‘a gift of traditional Chinese medicine to the world’. In the late 1960s, she had selected an extract of the sweet wormwood, Artemisia annua, from a screen of traditional Chinese medicines against the malaria parasite and then used the ancient medical literature as a guide to discover the active chemical ingredient. The compound in question – a sesquiterpene lactone named artemisinin – has now been the mainstay of antimalarial therapy for over a decade, and artemisinin-based combination therapies (ACTs) are currently estimated to save well over 100,000 lives each year in Africa alone. Yet resistance to ACTs is already growing: the ongoing ‘arms race’ against the malaria parasite requires a constantly replenishing pipeline of new drugs.

This arms race has a long history. The intermittent fevers that are characteristic of malaria have been recognised since classical times; it was endemic throughout medieval Europe and mentioned in eight of Shakespeare’s plays. The first effective antimalarial drug was quinine, extracted from the bark of the cinchona tree that had been known for centuries in its native South America as a remedy for fever.
Malaria is caused by single-celled parasites in the genus Plasmodium, which are carried by two hosts, Anopheles mosquitoes and humans. Five species can cause malaria, but two, P. falciparum and P. vivax, are by far the most important. There are subtle differences between their life cycles, but the basic details are the same. When an infected female mosquito – the disease can only be passed on by females – bites a human, parasites rapidly pass into the host’s liver cells where they become dormant and asexual; during this phase the patient has no symptoms.
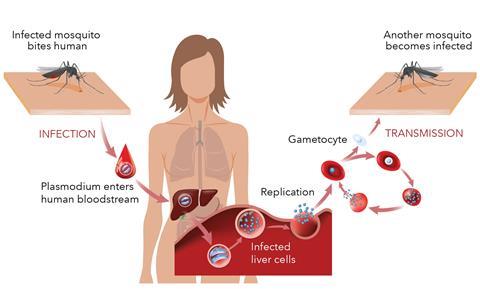
When liver cells rupture, any parasites they contain pass into the bloodstream and the red blood cells. There, they reproduce by approximately 10-fold every two days, at which point infected cells rupture releasing the new parasites; this leads to the clinical symptoms of fever and pain. A small number of newly-infected red blood cells have parasites that differentiate into male and female gametocytes, which are transmitted to the next mosquito that bites; they reproduce in the mosquito and the cycle begins again. Most antimalarials developed so far are active against only one of these stages: the blood stage, to cure current symptoms. However, to stop the disease spreading and ultimately aim for disease elimination, it is necessary to also target other stages of the lifecycle: the liver stage, to prevent transmission from mosquitoes or relapse of vivax malaria; and the gametocytes, to block transmission to mosquitoes. The over-arching goal of ‘malaria no more’ can only be achieved by simultaneously targeting every stage of this complex cycle.
Beyond quinine
Many new medicines entered the clinician’s armoury between quinine in the 17th century and artemisinin and its derivatives in the 21st, but resistance has always followed sooner or later. It was a combination of resistance to the insecticide DDT, used to kill mosquitoes, and to the once-effective antimalarial chloroquine that led to a widespread increase in deaths from malaria in the 1960s. The most recent WHO statistics show that over 200 million cases of malaria occur each year, over 90% of them in Africa. The death rate is low in otherwise healthy adults but much higher in young children: a child dies from malaria somewhere in the world every five minutes.
The pharmaceutical industry has for many years been reluctant to invest in treatments for infectious diseases, particularly those like malaria that are endemic in poor countries. At the turn of the millennium the overall malaria pipeline was particularly dry, with artemisinin and a few historical partner drugs seemingly the only options to prescribe. Today, however, the pipeline is much healthier, with a wider range of compounds under consideration at different stages, some are even being taken up by the pharma giants that were so dismissive a couple of decades earlier. And much of this positive ‘sea change’ in priorities can be put down to one not-for-profit Swiss foundation: the Medicines for Malaria Venture (MMV), now approaching its 20th anniversary.
‘We started in 1999 with support from four major public and charitable donors, and we now have 14,’ explains Jeremy Burrows, head of drug discovery at MMV. ‘Our focus is entirely on discovering, developing and delivering the next generation of small molecules to treat malaria, and by 2021 we will have invested over a billion dollars in novel drugs.’ No research is done at MMV’s offices in Geneva, although over half the staff located there are research scientists; instead, it acts as a ‘virtual pharma company’, supporting work through collaborative partnerships in a network of academic and industrial labs in 47 countries. Since 1999, 19 novel drug targets for malaria have been validated, and 12 new products developed with its partners have been registered in the last 10 years alone.
MMV funding is distributed to projects through a highly competitive annual call for proposals. ‘Every year, we look through our portfolio, decide where the most pressing research needs are, and structure the call accordingly,’ says Burrows. The current portfolio includes compounds that act at all points in the parasite life cycle. And unlike some not-for-profit drug discovery ventures, MMV is able to progress a compound, through partnerships, from discovery through clinical studies and all the way to regulatory approval and delivery to patients.
And MMV can also breathe new life into old projects yet to reach stringent regulatory approval. A case in point is the 8-aminoquinoline compound tafenoquine. It was originally synthesised in 1978 in the Walter Reed Army Institute of Research near Washington DC in the US as a potential prophylactic against P. falciparum infection but soon dropped. A collaboration between MMV and the pharma giant GSK resulted in it being taken on for development against recurrent P. vivax infection. Patients with this form of malaria can relapse years after the initial infection, and the disease is only considered cured when dormant parasites have been completely cleared from a patient’s liver. For decades another 8-aminoquinoline, primaquine, has been the only medicine available for this indication, but it is itself rapidly cleared from a patient’s body and a long course of treatment is necessary to achieve the ‘radical’ cure. ‘Tafenoquine has a much longer half-life in blood plasma than primaquine,’ says Gavin Koh from GSK in Uxbridge, UK. ‘This makes it amenable to administration in a single dose, which is much more convenient for patients.’

Koh was recently part of a large team, led by GSK’s JP Kleim, that has taken this drug successfully through a clinical trial that showed this single dose to be about as effective as a full course of primaquine in preventing relapse in patients with P. vivax infection. ‘We worked in six malaria-endemic countries in three continents, and many of the centres we used had little experience of clinical trials and very poor facilities,’ explains Koh. ‘But despite the logistical challenges, over 95% of patients completed the studies, with a massive effort by all concerned’.
Tafenoquine is the first new treatment for preventing relapse of P. vivax infection to have been licensed in over 60 years, but it cannot be used in all cases. Both these aminoquinolines cause severe side-effects in people with an inherited deficiency in the enzyme glucose-6-phosphate dehydrogenase. This affects about 8% of the population of many malaria-endemic countries, so the aminoquinolines are best prescribed after testing for this enzyme activity: this is a rare example of a specific safety test used for a tropical disease.
GM Mosquitoes
At least since the invention of DDT, the fight against malaria has been a two-pronged one, targeting the insect vectors of the disease as well as the parasites themselves. With controversies over toxicity surrounding insecticides, scientists are increasingly turning to sophisticated biological methods of vector control. Andrea Crisanti and his team at Imperial College London, UK, have developed a technique to genetically modify Anopheles gambiae mosquitoes to make the females sterile.
‘We use Crispr–Cas9 to target a mosquito gene called doublesex that controls sex differentiation,’ explains Crisanti. ‘This introduces a modification that destroys the female form of the gene, so female mosquitos develop into completely sterile hermaphrodites.’ Without fertile females, a relatively small population of mosquitoes in a closed cage will collapse rapidly. ‘The next state will be to reproduce the results in an environment that is much closer to the natural one, in large cages in a field site in Italy where it is hot enough in the summer for Anopheles to breed,’ Cristiani adds.
A decade of development
The Drug Discovery Unit (DDU) at the University of Dundee in Scotland has been developing novel drugs for malaria since 2008. It was set up by two leading parasitologists, Alan Fairlamb and Mike Ferguson with one aim: the discovery of novel small molecules to treat neglected tropical diseases. ‘We now employ about 100 people and our labs are better equipped than those of many biotech companies of a similar size,’ says Julie Brady, business development manager at the DDU. ‘We can apply for capital development as well as research grant funding, and so have the best of both worlds.’

The unit’s malaria programme receives much of its support from MMV. Its most notable success to date has been a recent one: a candidate molecule that is now being taken into clinical trials by Merck KGaA. This compound, originally known as DDD107498 and now, only marginally less forgettably, as M5717, was discovered through a phenotypic screen of a library of a few thousand kinase-inhibitor-like compounds. This type of screen pre-supposes nothing about a compound’s mechanism of action, simply testing its ability to kill the parasites at various stages of their life cycle. In this case, the screen identified 11 interesting chemical series of which seven were already in MMV’s portfolio, so the Dundee group retained four. ‘The fourth series that we investigated, which was based on a quinoline-4-carboxamide scaffold, has proved the most interesting,’ says the DDU’s head of chemistry, Ian Gilbert. ‘Optimising the initial hit in this series gave us a compound that was able to kill parasites in multiple life stages, that was active in a mouse model of malaria and that had good pharmacokinetics.’
The next step was to find out the compound’s mechanism of action, and this used a method that has only been feasible since sequencing of complete eukaryotic genomes became routine. The Dundee group’s collaborators at Columbia University Medical Center in the US cultured P. falciparum in the presence of the compound until resistance developed, and then the genomes of the resistant parasites were sequenced at the Wellcome Sanger Institute near Cambridge, UK. All resistant parasites shared mutations in the gene that codes for a protein called translation elongation factor 2 (eEF2). ‘This protein is one of those that is essential for protein synthesis: when the drug binds to eEF2 this process stalls and the parasites die,’ says Marcus Lee, a parasitologist at the Sanger Institute. However, anti-infective drugs that inhibit protein synthesis – such as many successful antibiotics – rely for their success on there being enough difference between the molecular machinery for protein synthesis in the pathogens and its human equivalent for human protein synthesis to be unaffected. ‘We know less about how this evolutionarily conserved process works in Plasmodia than we do in humans or bacteria, but toxicology studies of this compound suggest that selectivity for the parasite eEF2 over the human enzyme is achievable,’ says Lee.
Open season
The step change in investment in antimalarials that MMV has led in the last 20 years has been extremely welcome, but the projects that it funds often look like conventional drug discovery ones. Matthew Todd, professor of drug discovery at the UCL School of Pharmacy in London, UK, is pioneering a more radical approach. The Open Source Malaria consortium, founded when he was based at the University of Sydney in Australia, aims to ‘do drug discovery differently, with no secrecy and therefore with no patents’. Todd took his inspiration from open-source software projects like Linux and Wikipedia, and set up a completely open project in which all data generated is immediately put in the public domain and which anyone, regardless of their credentials, can take part in. ‘We started with the library of over 13,000 small molecules that had been released into the public domain by GSK under a Creative Commons license,’ explains Todd. Other compound libraries have since been made publicly available and added to the project. A large community of research groups, individuals and even a few school science departments submit their results in open lab notebooks, with all decisions made through consensus in virtual lab meetings.
We started with the library of over 13,000 small molecules in the public domain
Todd and his many co-workers have validated and explored four promising compound series since the project began in 2010. Three were originally derived from molecules in the first GSK dataset, and the fourth originated in a dataset that Pfizer released into the public domain before closing its UK research base in Sandwich. The first series to be investigated split into two sub-groups, one containing an essential but labile ester group and the other showing high potency but low solubility; the second was parked when the collaborators realised that another group was working on it. ‘We are now concentrating on molecules from series three and four,’ says Todd. ‘Interest in the third series stems largely from its suggestion of a completely new target in one of the parasite’s tRNA synthetase enzymes, and some of the fourth set have shown antimalarial activity in a mouse model yet remain in the public domain.’
These case studies illustrate how the constant arms race between scientists and malaria parasites has shifted in our favour in the last 20 years. There are many promising antimalarial compounds out there, and some mechanisms and funding to take them through development are finally in place, but we will still need more: particularly if, as some suggest, the parasites’ endemic range increases as the climate changes. And both open-source drug development and big pharma – and everything in between – will have roles to play in achieving MMV’s vision of making malaria no more.
Clare Sansom is a science writer based in London, UK











No comments yet