
‘There’s simply no other reaction in chemistry that will functionalise that carbon–hydrogen bond,’ says John Hartwig from the University of California, Berkeley, in the US about his team’s discovery: a catalyst that tackles the least reactive bond in a molecule. The reaction replaces the strongest primary C–H bond with a boron group.
‘I think it opens up the area of C–H functionalisation to another level,’ says Varinder Aggarwal, an expert in borylation chemistry at the University of Bristol, UK, who wasn’t involved in the work.
Many C–H functionalisation catalysts work on a molecule’s most reactive bonds, while others need a directing group to guide it to a specific position. Back in 2000, Hartwig’s team found a catalyst that preferred primary C–H bonds above all others. However, the reaction required a huge excess of the substrate, which is impractical if the compound is expensive or the product of a lengthy synthesis.
Now the chemists designed an iridium catalyst that borylates the most stable, least reactive aliphatic C–H bonds in a molecule. The catalyst’s reaction rate is almost two orders of magnitude higher than that of similar catalysts, which allowed the team to use only one equivalent of the substrate for the first time.
The catalyst’s selectivity is particularly apparent in dehydroabietic acid, says Hartwig. The compound that has multiple aromatic and benzylic C–H bonds as well as aliphatic secondary and tertiary C–H bonds. But the catalyst only goes after a single methyl hydrogen transforming it into a boron group, albeit in moderate yield.
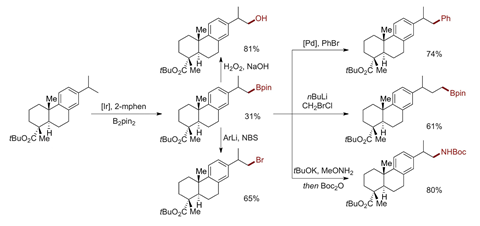
If the substrate doesn’t have CH3 groups, or if they are blocked by steric hindrance, the catalyst functionalises the least activated secondary C–H. In saturated heterocycles, this is the position beta to the heteroatom, which ‘is typically inaccessible by classical chemistry’, Hartwig explains.
The fact that the reaction is a borylation is an added bonus, says Aggarwal. ‘Boron is probably the most versatile functional group in organic chemistry. Being able to take simple, unreactive alkyl groups, carry them through a synthesis and then convert them into a carbon–boron bond is really, really useful because the C–B can then be transformed into a whole host of other functional groups.’
However, the catalyst’s unusual activity can also be problematic: ‘What solvent do you do the reaction in?’ wonders Aggarwal. ‘Trying to find a solvent that does not undergo the reaction itself is challenging.’ Hartwig’s team uses cyclooctane, a non-polar solvent that many molecules don’t dissolve in. ‘We definitely will need to come up with a solvent that is more polar, but still resistant toward this type of C–H activation chemistry,’ Hartwig says.
Exactly why the catalyst chooses the toughest bonds over all others is still unclear. While Hartwig’s group is working on uncovering the full catalytic cycle, he hopes that the work will encourage others to go after those challenging, unreactive C–H bonds. ‘I wonder if it’s a little bit like a four-minute mile; once you know it can be done, then there become many different solutions to it, and different people can meet that goal.’
References
R Oeschger et al, Science, 2020, 368, 736 (DOI: 10.1126/science.aba6146)














No comments yet