Approach can construct wide range of new antibiotic candidates from simple chemical building blocks
A new fully synthetic way to make a class of antibiotics called macrolides from simple building blocks is set to open up a new front in the fight against antimicrobial drug resistance.1 The US team behind it identified molecules with potent antibiotic activity against bacterial strains that are currently resistant to drugs, which suggests the approach could lead to new macrolide antibiotics and help combat the growing global health threat of antibiotic resistance.
For decades, new antibiotics have been discovered and made using a process called semi-synthesis, which chemically modifies natural products. However, the snag is that it’s difficult to selectively control the desired chemical modifications on structurally complex natural products, which greatly limits the new drug candidate molecules that can be made.
Erythromycin, a macrolide antibiotic obtained by fermentation, was first discovered in 1949 and its semi-synthetic derivatives azithromycin and clarithromycin are commonly prescribed for treating various respiratory tract and skin infections such as pneumonia, gonorrhoea and streptococcal pharyngitis. They work by disrupting the bacterial ribosome and hence protein production. But bacterial resistance to these approved macrolide antibiotics is now widespread and semi-synthesis can’t provide the new macrolides required to keep up.
‘Our goal was not to make a single molecule [as with semi-synthesis] but to develop a design strategy that by its nature would enable the preparation of thousands or even tens of thousands of clinical candidates,’ explains Andrew Myers at Harvard University, US, whose group led the study. ‘With a fully synthetic route we have access to orders of magnitude greater numbers of structures for study, and can effectively modify just about any position of the scaffold. That can’t be said of semi-synthetic approaches.’
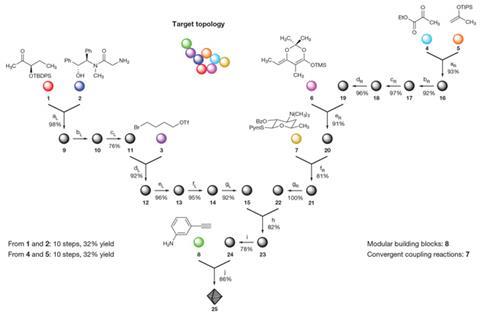
Myers’ lab previously developed the first practical, scalable synthetic route to make tetracycline antibiotics, also based on building blocks, in 2005.2,3 This time, the group turned to macrolides which involved using eight different building blocks to assemble large 14 to 16 membered rings by a series of convergent coupling reactions; constructing these macrocyclic rings via synthetic means has traditionally been a tough challenge.
Candidate diversity
What Myers’ team did was invent crucial but simple chemistry to enable the necessary bond formations, including an aldol addition reaction – a type of carbon–carbon bond-forming reaction – which is a key early coupling reaction in the route. ‘What we did not do is almost as important as what we did do,’ adds Myers. ‘We vetoed any plans that were overly long and linear, or which used chemistry that would not be amenable to scale.’
[[At the outset of the project I never dreamed that our solution would be as powerful or general as it turned out]]The method produced over 300 structurally diverse macrolide antibiotic candidate molecules previously inaccessible via semi-synthesis. These included the approved drug telithromycin, and the clinical candidate solithromycin, which are effective against community-acquired pneumonia.
What’s more, gram quantities of macrolides were made which is ample for drug discovery purposes. Tests revealed that most of the compounds had antibiotic activity against an array of pathogenic bacteria, and some were active against bacterial strains that show resistance to macrolides in current use. These included the clinically relevant ‘superbug’ strains methicillin-resistant Staphylococcus aureus, better known as MRSA, and vancomycin-resistant Enterococcus.
‘What is particularly remarkable here is that this class of natural products has been explored extensively in the context of semi-synthesis, synthetic biology and even total synthesis yet this new approach is the only one that is robust and flexible enough to provide a myriad of new, deeply modified analogs in an economically viable fashion,’ comments organic synthesis researcher Phil Baran from The Scripps Research Institute, La Jolla, US. ‘Since overcoming resistance is a constant battle, the value of this approach is that it offers an opportunity to access new areas of relevant chemical space in a significantly truncated timeframe.’
‘Frankly, at the outset of the project I never dreamed that our solution would be as powerful or general as it turned out,’ says Myers. ‘One reason for optimism is that the chemical space we can explore is so vast that it will take years if not decades to thoroughly investigate it.’
References
1 I B Seiple et al, Nature, 2016, 533, 338 (DOI: 10.1038/nature17967)
2 M G Charest et al, Science, 2005, 308, 395 (DOI: 10.1126/science.1109755)
3 M G Charest, D R Siegel and A G Myers, J. Am. Chem. Soc., 2005, 127, 8292 (DOI: 10.1021/ja052151d)









No comments yet