
Upregulated gene triggers laminin-α1 protein production, reviving paralysed limbs
Researchers in Canada and Sweden have used a novel Crispr system to reverse harm caused by muscular dystrophy – usually thought to be permanent – in mice. Pre-clinical studies often treat animals early, before symptoms can be detected, says Ronald Cohn, senior scientist, president and chief executive officer of The Hospital for Sick Children in Toronto.
‘Our results now show that the therapeutic window may indeed be wider,’ Cohn tells Chemistry World. This is significant as all patients ‘are currently diagnosed at a time when symptoms are already presenting’, he adds. The researchers are now exploring partnerships with industry to work towards bringing the research into a clinical trial.
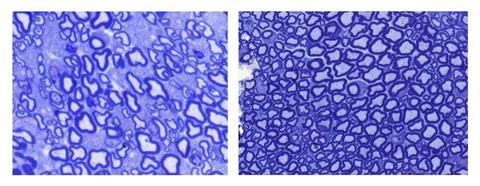
There are many forms of muscular dystrophy, each resulting from a mutation in a gene that tells the body to make an important muscle protein. The version this treatment targets, MDC1A, arises with versions of the protein laminin-α2 that don’t work properly. MDC1A affects around one child in 30,000, often causing muscle weakness from birth that means many sufferers cannot walk. After puberty, they may develop life-threatening breathing problems.
One gene to help them all
Current treatments for MDC1A only slow the disease’s progression. Researchers looking for better treatments often target the problematic mutation. However, this brings practical challenges, as there are many possible mutations, so small patient groups with each mutation would each need tailored treatments.
Madeleine Durbeej from Lund University in Sweden has been studying a different way to deal with the loss of laminin-α2. Her approach exploits the fact that MDC1A sufferers have working versions of a gene called LAMA1 that can tell cells to make a similar protein, laminin-α1. ‘Upregulating’ production of that protein should then enable it to substitute for laminin-α2.
Crispr approaches are best known for offering a ‘satnav and scissor combo’ for finding a genetic sequence and cutting it. Upregulating the LAMA1 gene involves blunting the scissors, mutating the Crispr-associated protein 9 (Cas9) so this enzyme can’t cut. Together, Cohn, Durbeej and colleagues also attach their nuclease-deficient Cas9 (dCas9) to four copies of a protein called VP16 that can upregulate laminin-α1 production.
Small and stealthy
The researchers also had to smuggle these proteins across the cell wall border before their Crispr-dCas9 satnav can access the genes they’re aiming for. Adeno-associated viral vectors (AAV) have become a regular choice for such smuggling, including in gene therapy, as they don’t set off adverse immune system responses. However, they come with a significant drawback: They can only carry relatively small cargos. Cohn and Durbeej’s team therefore switched to Cas9 from Staphylococcus aureus, which is much smaller than the usual Cas9 that researchers use, calling the combined system SadCas9.
The scientists tested their AAV-borne SadCas9 in MDC1A mice unable to move their hind legs when born. After three weeks the researchers gave four groups of three mice differing treatment doses and forms. By seven weeks the mice given the highest doses of the form including RNA molecules to guide SadCas9 to its target could move their hind legs. ‘We demonstrated that CRISPR can sufficiently upregulate a disease-modifying gene in a mutation-independent approach to correct the phenotype of mice with MDC1A,’ Cohn tells Chemistry World.
Kate Adcock, Director of Research and Innovation at Muscular Dystrophy UK, calls the study ‘an exciting piece of research’, which should give hope to people with MDC1A and their families. ‘There is still a way to go before any treatment is brought to clinic,’ she adds. ‘The next step will be to replicate the study in other animal models before we extend the study to humans.’
Markus Ruegg from Biozentrum, University of Basel, Switzerland, notes that this study replicates earlier findings that laminin-α1 can reverse the effects of MDC1A. However, as Lama1 is too big for gene therapy something like the SadCas9 approach is needed to produce a treatment, he adds. However Ruegg cautions against being overwhelmed by the prospects of such novel ideas. ‘It really is a very nice proof-of-concept study but not more,’ says.
References
D U Kemaladewi et al, Nature, 2019, DOI:10.1038/s41586-019-1430-x















No comments yet