
Chemists have cracked the decades-old mystery of the mirror-symmetry breaking Soai reaction, one of the strangest and most unique transformations in organic chemistry. It turns out that an enzyme-like mechanism allows tiny chiral imbalances to create single enantiomer products from non-chiral starting materials. Understanding the Soai reaction might provide clues to how life came to be built on DNA, amino acids and sugars that exist almost exclusively in one mirror image form.
The reaction between pyrimidine-5-carbaldehyde and diisopropyl zinc was discovered in 1995 by Kenzo Soai from the Tokyo University of Science in Japan. The alkylation creates a chiral alkoxide, which acts as its own enantioselective catalyst. ‘Starting with tiny amounts of a catalyst, the reaction can amplify to almost enantiomeric purity,’ explains Scott Denmark from the University of Illinois, Urbana in the US. ‘The phenomenon had been predicted in the 1950s by [theoretical physicist Frederick Charles] Frank as a mechanism by which the Earth became biologically homochiral.’
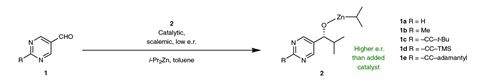
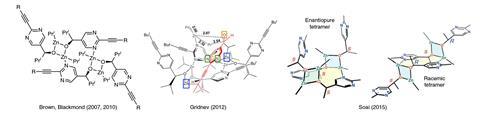
The reaction has a number of idiosyncratic substrate requirements: it only works with diisopropyl zinc and a pyrimidine-5-carbaldehyde. Although chemists had found a tetrameric product aggregate that was most likely the catalytic species, nobody had come up with a binding model and transition state structure to fully explain the reaction’s mechanism.
Denmark’s team now combined experimental and computational studies to make sense of the reaction’s strangely stringent conditions. ‘We found the first example that broke the mould of the substrates that would do this reaction,’ says Denmark. Instead of a pyrimidine, the team used a pyridine derivative, which is more soluble and easier to study by nuclear magnetic resonance.
The Soai reaction works with diisopropyl zinc, but not with diethyl zinc. The latter, it turns out, forms an aggregate that is too stable and catalytically dead. ‘Zinc alkoxides naturally want to be cubic,’ explains Denmark. The Soai zinc alkoxide product is too sterically hindered to form cube-shaped aggregates. As it breaks the cube, the nitrogen swings around and picks up zinc’s empty coordination site, forming the catalytically active tetramer. ‘The cube escape forms a species that has a pocket that is uniquely designed to bind itself because its spacer is itself,’ Denmark says.
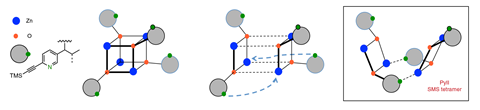
Denmark and colleagues also discovered the secret to the reaction’s asymmetric amplification power. Surprisingly, both the racemic and the enantiopure tetramer can act as a catalyst. But the pyridine is extremely unlikely to bind to the racemic catalyst as this is very energetically unfavourable. Only binding to the enantiopure tetramer creates a favourable two-point interaction. This, Denmark points out, is akin to classic Michaelis–Menten enzyme kinetics.
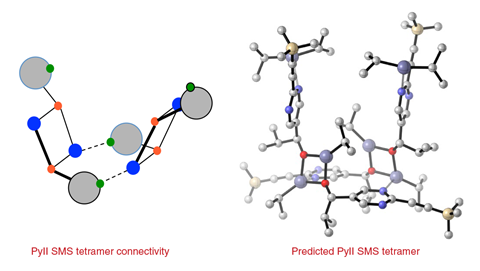
Soai says that the team’s synthetic and spectroscopic experiments had him convinced, although he didn’t want to comment on the computational parts of Denmark’s study. However, he points out that ‘the aggregation state of catalyst may change during the reaction according to the concentration of catalyst’. ‘I think their result constitutes the kick-off of the study of the transition state of the reaction,’ Soai adds.
‘Unfortunately, we had to conclude that this system is just so unique that we don’t know if there are general lessons to take away from it to build other autocatalytic systems,’ Denmark says. But his team is not finished with the reaction yet. ‘Most importantly, we want to learn how the product, after the isopropyl group is delivered, reorganises to form empty tetramers that can pick up another substrate.’
References
S V Athavale et al, Nat. Chem., 2020, DOI: 10.1038/s41557-020-0421-8















No comments yet