Temperature can severely influence the conformational state and ligand binding properties of a protein, new research shows. Consequently, computational modelling based solely on cryogenic structural data may produce misleading results.
Nearly all crystallographic datasets collected for determining protein structures are obtained at cryogenic temperatures. The protein crystals are cryocooled with liquid nitrogen, and when a crystal is bombarded with x-rays, the low temperature conditions help to limit x-ray damage, which allows users to collect high-resolution datasets with high completeness. Although it can create more crystal damage, it is also possible to conduct experiments at room temperature. Many proteins are flexible so have dynamic structures, and certain conformational states might only be accessible at room temperature. Users may therefore not observe, or correctly capture, the dominant conformational state of a protein at natural biological temperatures when studying a protein under cryogenic conditions. In addition, if multiple protein conformations are possible, then these may arise in different proportions at different temperatures.
Now, Marcus Fischer of St Jude Children’s Research Hospital in Tennessee, US, and colleagues have performed the first systematic analysis of temperature artifacts in protein structures. ‘People may be overinterpreting data that are actually cryo-artifacts. When you look at only cryo- and start optimising … you may be misled. You just don’t know,’ he says.
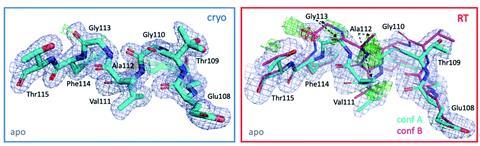
The team began their study by looking at the model system T4 lysozyme L99A, which has more than 700 structures deposited in the Protein Data Bank (PDB). However, upon collecting the largest temperature series to date of nine dataset pairs at both cryo- and room temperature, Fischer’s team observed a new apo helix confirmation that is relevant to ligand binding. ‘That’s what we’re advocating here,’ says Fischer. ‘Room temperature can provide additional insights into the conformational landscape that can be explored for ligand design and discovery.’
Computational modelling is essential for modern drug discovery and design, and for ligand docking studies to translate to successful drug discovery, it is crucial for the starting protein model to be as true to the in vivo target as possible. By collecting 14 new high-resolution datasets and combining these with datasets from the PDB, the team could analyse nine pairs (at cryo- and room temperature) of the T4 lysozyme L99A structure. They found that more than a third of residues, most located near the ligand binding site, responded to temperature changes. They also examined ligand binding and occupancy over hundreds of datasets, which revealed new ligand binding poses at room temperature. The investigation was extended to four other protein classes and clearly demonstrates that room temperature structures can provide insights that are not captured with cryogenic structures. Using the collected data as a starting point for modelling studies demonstrates that alternate features can, and perhaps should, be carefully modelled rather than discarded as minor states.
‘A structure is a snapshot’
‘Those who base future experiments on crystallographic results should be aware of the limitations,’ comments Eleanor Dodson, an emeritus professor at the University of York, UK, and an expert in computational modelling of protein crystallography. ‘Results will be influenced by temperature, crystallisation conditions, radiation damage, etc. A structure is a snapshot. Good modelling tools should take these results on board, but computational modelling is also having to deal with approximations.’
Cryoprotection pioneer Elspeth Garman, at the University of Oxford, UK, says developers of computational modelling tools should take notice of this study: ‘PDB cryo-structures will not be as productive a training set as room temperature-structures would be. Due to the development of serial synchrotron crystallography and reliable data analysis software to merge data from many different crystals, room temperature-crystallography is undergoing a renaissance, which will expand the number of room temperature-structures in the PDB and enlarge the room temperature-structure training set for AI-based computational modelling tools.’
While this research highlights the drawbacks of relying on cryogenic structures, Keith Wilson, an expert in methods development for protein crystallography at the University of York, UK, says that room temperature data collection is not always possible when studying biologically active macromolecules: ‘For most proteins, room temperature data collection gives very rapid crystal death and a large number of crystals are required to record a complete set of data. Room temperature data collection, including in-plate situ techniques, have been applied to crystals for which cryogenic freezing has proved intractable such as a number of membrane proteins. But this does again mean a large number of crystals are required for complete data. I would see cryogenic data collection as continuing to be the norm for the vast majority of projects. It is just too challenging to record room temperature data for most studies’
However, Fischer is keen to encourage more researchers to try room temperature experiments. ‘There’s not much to lose other than time and maybe a little bit of money, but the insights that you gain – it’s almost a new experiment. You just get so many new, different insights that it is worth a shot.’
References
This article is open access
S Y C Bradford et al, Chem. Sci., 2021, 12, 11275 (DOI: 10.1039/d1sc02751d)












No comments yet