After hundreds of failed drug candidates, James Mitchell Crow asks if this is the last roll of the dice for the amyloid theory of Alzheimer’s disease
When Europeans crossed the Atlantic to reach the New World in the 16th and early 17th century, the deadly diseases that travelled with them included smallpox, measles and yellow fever. To that list, now add Alzheimer’s disease. According to a recent genetic study, a single Spanish conquistador is the probable source of a global hotspot for Alzheimer’s cases in Antioquia, northern Colombia.1
Antioquia has the world’s largest population of people with familial Alzheimer’s, the heritable early-onset form of the disease. Geneticists traced the faulty gene responsible back to an individual of European origin who lived during the early 17th century – the height of the Spanish conquest.
Of the conquistador’s 5000 or so descendants alive today, approximately 1500 carry the mutated gene that destines them to develop the disease. In the general population, Alzheimer’s typically strikes the over-65s. For the extended Columbian family, the average age of disease onset is just 45 years. Memory loss is the first outward sign, followed by behavioural and personality changes. Language ability is progressively lost. Movement disturbances and seizures characterise the latter stages of the disease. In this family’s case, death typically follows just eight years after symptoms appear.2
Every drug since 2002 has failed – that’s a few hundred compounds
So far, all attempts to develop a drug for Alzheimer’s have failed. But could the very predictability of familial Alzheimer’s provide the path to a treatment? After many failed drug trials, most Alzheimer’s researchers now suspect the only way to treat the disease is to start very early, in symptom-free subjects whose brain is yet to suffer significant damage. Familial cases, where individuals develop the disease very predictably, allow us to put that idea to the test.
The Columbian family are willing participants of one such trial. Researchers at Washington University in St Louis in the US, who head the Dominantly Inherited Alzheimer Network (DIAN), lead another trial incorporating several families. If the trials succeed, it could pave the way for the first effective Alzheimer’s treatment – while also corroborating our leading theory of the underlying molecular cause of the disease.
If the early-intervention trials fail, however, then that theory essentially fails with them.
16 years of hurt
The intense focus on drug trials with early-onset Alzheimer’s cohorts reflects the fact that, in drug trials involving patients with the far more common non-familial, late-onset form of the disease, all attempts at treatment have failed.
The demand for a drug is great. Familial Alzheimer’s represents only around 1% of cases; the far more common form, sporadic Alzheimer’s, can affect anyone. It is the most common form of dementia, and among the elderly it is the third most common cause of death after heart disease and cancer.3
Countless resources have been thrown at trying to understand and treat the disease. Merck and Co, Eli Lilly and Pfizer are among the big players who have invested heavily in Alzheimer’s drug research and development. But not one of their attempts has slowed the disease’s progression in clinical trials. ‘Every drug since 2002 has failed – that’s a few hundred compounds,’ says Andrew Doig, who researches the disease at the University of Manchester in the UK. The rare successes prior to 2002 offer only temporary relief of some symptoms and do not slow progression of the disease.
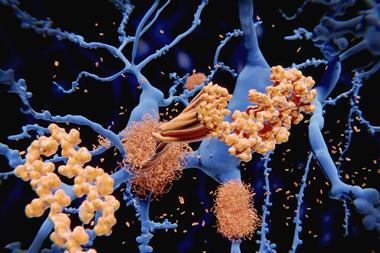
Most of these attempted therapies have revolved around the amyloid cascade theory of Alzheimer’s disease. The theory grew from the post-mortem analysis of the brains of Alzheimer’s patients, and the hallmark features found there called plaques. 34 years ago, a young biochemist called Ralph Martins was part of an Australian study to identify what these plaques consisted of. Drug development was a key part of the motivation.4 ‘If we knew there was a particular protein in these structures, then we had something to target,’ says Martins, who now leads Alzheimer’s research groups at Edith Cowan University in Perth and at Macquarie University in Sydney.
‘The abundant protein that we found in the plaques, and that the Americans found in the blood vessels of the brain, consisted of a small protein everybody now knows as beta-amyloid,’ Martins says. ‘This has been a central aspect of our approach.’
The more that researchers studied amyloid beta, the more the pieces of the puzzle seemed to fall into place. In most cases of familial Alzheimer’s, the inherited faulty gene causes an excess of amyloid beta to accumulate in the brain. People with Down’s syndrome, who also develop amyloid plaques early in life, have an extra copy of the gene that codes for the parent molecule, amyloid precursor protein. ‘The brains of people with Downs have amyloid even at the age of five or six,’ Martins says.
Although numerous changes have been shown to occur in the Alzheimer’s brain, amyloid’s position as the kingpin protein of the whole process appeared set. The theory that emerged is that first you get too much beta amyloid in the brain, which eventually aggregates. That causes cellular effects like chronic inflammation and oxidative stress. A second protein called TAU is affected: rather than doing its job of stabilising the neurons’ cytoskeleton, it also aggregates into tangles. After that – typically a decade or two after amyloid first began accumulating in the brain – neuronal cell death and the first symptoms of dementia follow.
If the amyloid cascade theory is correct, then the way to intervene appears obvious. Interfering with the excess production and aggregation of amyloid should halt the whole process. Two strategies for interrupting amyloid aggregation that, in lab tests at least, look like particularly good therapeutic options have been developed.
One possibility is to block the activity of an enzyme in the brain – a beta secretase enzyme called BACE 1 – that cleaves the amyloid precursor protein to produce amyloid beta. Merck & Co, AstraZeneca and Janssen are among the pharmaceutical companies that have developed and run large clinical trials on small-molecule beta secretase inhibitor drugs to block the activity of the enzyme.5
The other highly anticipated strategy was to develop monoclonal antibodies that recognise and mop up beta amyloid in the brain, preventing plaque formation and promoting clearance of the problem peptide from the brain. Pfizer, Biogen and Eli Lilly are among the pharma firms to have developed prospective Alzheimer’s therapies in this category.
In both cases, cell studies and animal models suggested all these therapies were safe and effective – they did what they were designed to do. Early stage clinical trials with humans appeared to offer glimmers of promise. And yet for every final stage clinical trial completed so far, the drugs have failed to show any slowing of participants’ cognitive decline.
In February 2018, Merck discontinued the Phase III trial of its BACE 1 inhibitor verubecestat (MK-8931). One month earlier, Pfizer threw up its hands and quit Alzheimer’s research altogether following its own failures with its bapineuzumab antibody.6
Why aren’t these drugs working? Right now, nobody is certain. But there are a number of theories. And a handful of drug trials going on right now could soon tell us which one is right.
Amyloid ambiguity
One possible reason for the endless string of drug trial failures is that the amyloid theory of Alzheimer’s – a hypothesis so many scientists have been seduced by – is wrong. ‘The failure of all those clinical trails tells us what it’s not, and that’s very important information,’ says Ashley Bush, an Alzheimer’s researcher and clinician at the Florey Institute of Neuroscience and Mental Health in Melbourne, Australia. The amyloid theory ‘has been flogged very hard, and if it had something to say surely it would have said it by now’. At the very least, it’s about time other theories received more oxygen, he argues.
For the past 25 years, Bush has studied the role of metals and oxidative stress in neurodegeneration and dementia. Bush’s research points to elevated iron levels in Alzheimer’s patients’ brains, not amyloid, as the most significant factor at play – and he currently leads a clinical trial to test iron-clearing drugs in Alzheimer’s patients (see box Iron in the fire below). Iron, which can readily switch between the +2 and +3 oxidation state, is renowned for generating free radicals.
The amyloid theory has been flogged very hard, and if it had something to say surely it would have said it by now
Doig also has doubts over the amyloid theory – although not so grave as Bush’s. For Doig, it is the cascade aspect of the theory that doesn’t ring true. ‘The idea it is literally a linear cascade – I think this is wrong, and what you get is lots of feedback loops.’ The onset of oxidative stress and inflammation are not only caused by amyloid aggregation, but they also feed back and induce more aggregation, for example. As a result, drugs that hit only amyloid may be ineffective. ‘If you just hit one point, the system can bypass it and it wouldn’t have an effect. You want to hit multiple points in the pathway to decrease the overall flux through the pathway.’
Complicating efforts to pin down amyloid’s role is that it is an exceptionally slippery substance to handle.7 Small, soluble oligomers of amyloid beta are thought to be the most toxic form of the peptide, says Doig. The trouble is, there’s a whole zoo of them. ‘They can be dimers, trimers, hexamers, dodecamers – we know there is five different kinds of hexamer, for example – and you can’t purify any of them,’ he says. The mixture is highly concentration-dependant, so if you try to isolate one oligomer it re-equilibrates and forms another population. There is also more than one form of amyloid beta peptide. ‘You can have various lengths, they can have various chemical modifications. They all interact with each other, bind to each other, and perturb the aggregation and toxicity.’
As a result, researchers have often taken a step back and simply tested their therapies on cellular and animal models of the disease. But these too have been flawed, says Doig. Until recently, the cells typically used were aggressively growing cancer cell lines, which researchers would heavily dose with amyloid – at concentrations far higher than seen in the brain – to make them sick. ‘You’re hoping that adding these high concentrations and waiting a few days mimics the brain of a human aged 80 living with beta amyloid pathology for maybe 30 years at much lower concentration.’
Animal models have had similar problems, Doig adds. Mice genetically engineered to produce high amyloid beta levels certainly experience the memory impairment, but key aspects of the disease are missing – not least the fact their life span is unchanged.
Stem cell science offers a promising alternative. Fibroblast cells from patients with familial Alzheimer’s, complete with their faulty amyloid beta production gene, can now be converted into neurons – which get sick because they naturally produce too much amyloid beta. ‘This is a far superior model,’ says Doig. ‘We are not adding any extra amyloid beta, and these cells naturally show all sorts of pathological problems,’ he says. ‘This technology wasn’t possible five years ago.’
Doig hopes these new models will be a better screen for identifying hits in potential drug libraries, including drugs already approved for other conditions.
Iron in the fire
One of the most revealing studies of sporadic Alzheimer’s disease has been a longitudinal study, started in 2006, called the Australian Imaging Biomarker and Lifestyle Study (AIBL). Over 1000 people aged over 60 have been tracked for over a decade, and tested every 18 months. The study, still ongoing, is the most comprehensive study of cognition, imaging and measurement of blood-based biomarkers in the world.
Several striking observations have come from the trial, says Ralph Martins, one of its founders who is an Alzheimer’s researcher at Edith Cowan University in Perth and at Macquarie University in Sydney. For example, lifestyle factors such as a good diet and exercising regularly seem to be just as important for staving off dementia as they do for avoiding heart disease and diabetes. Another observation is that inflammation seems more important, and to kick in much earlier in the disease cascade, than previously thought. ‘I think in the future we will see combination approaches of amyloid lowering and drugs that target inflammation,’ says Martins.
Another AIBL insight with potential therapeutic applications is the significant role of iron. In one cohort of participants, a recently developed form of MRI called quantitative susceptibility mapping was used to chart iron levels across participants’ brains. The same patients received PET scans to measure amyloid plaque formation.11 As previous research has shown, amyloid levels do not accurately predict when symptoms will appear. ‘The addition of the second scan, that measured iron, allows us to be far more accurate,’ says Ashley Bush from the Florey Institute of Neuroscience and Mental Health in Melbourne, Australia, who was part of the study. ‘Subject to it being replicated in other cohorts, I think it was a pretty powerful improvement on the amyloid scan.’
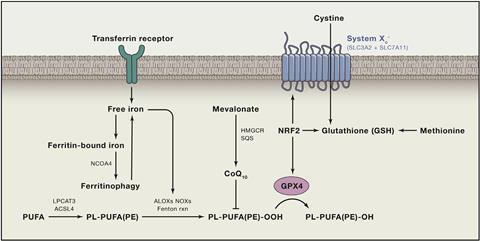
But Bush thinks the iron discovery is far more than useful in disease prediction. It is another piece of evidence that suggests iron, not amyloid, is the key toxic agent in the disease. ‘We can demonstrate the accumulation of iron causes a toxicity, and the mechanism of that we think comes from a form of programmed cell death called ferroptosis,’ he says. Like the similar-sounding apoptosis, ferroptosis is a mode of programmed cell death, first described by cancer researchers in 2012. Elevated iron levels induce the formation of lipid hydroperoxides, which kill the cell.12 The body uses iron accumulation to trigger ferroptosis to terminate malignant cancer cells. ‘We became intrigued because the chemistry resembles changes we see in Alzheimer’s post mortem tissue, namely lipid peroxidation and iron elevation,’ says Bush. ‘We have been examining whether the conditions in the brain inappropriately activate ferroptosis – it is a miss-sensing of an infection or invasion of microbes or some form of malignancy.’ In other words, iron accumulation in the brain in Alzheimer’s is triggering neurons to kill themselves.
Not everyone is yet convinced iron is the primary culprit, says Martins. ‘But it is something that deserves investigation,’ he agrees. Bush is already putting the idea to the test. Helpfully, an iron-lowering compound called deferiprone already existed, and has been tested in Parkinson’s. Bush is now leading the 3D (Deferiprone to Delay Dementia) study to test the drug in Alzheimer’s disease.
Unlike some of the latest amyloid-targeting drug trials, which are focused on intervening before the first symptoms appear, the 3D trial will include people already experiencing dementia symptoms. ‘We think this has got potential to work once a person has got early symptoms,’ says Bush. ‘I wouldn’t say ferroptosis is a major theory yet,’ he adds. ‘But there is a minority who are very excited by it.’
Too little, too late
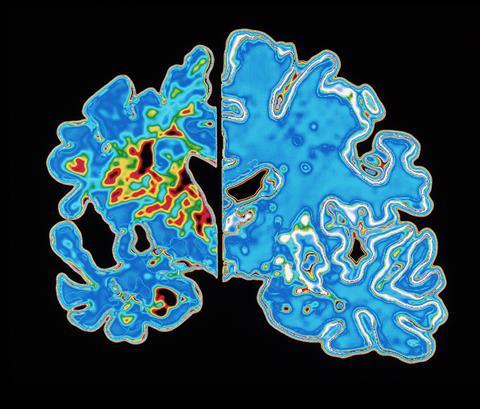
Perhaps the most popular theory for all the failed drug trials so far is that we’re using the drugs too late, on brains beyond repair. To intervene earlier, the issue is diagnosis. You don’t need a degree in neuroscience to notice something strange about the brain of an individual who has died of Alzheimer’s disease. The brain is shrunken, wizened, and clogged with deposits of beta-amyloid and tangles of TAU. Until recently, however, a post-mortem examination to check for amyloid plaques was the only way to confirm a patient’s dementia was Alzheimer’s disease.
During life, the best doctors could do was a memory test. ‘They might give you 12 objects on a tray to look at for a couple of minutes, take the tray away and 5 minutes later ask how many of those objects you remember,’ says Doig. A healthy person would remember 10 or 11 objects. Alzheimer’s patients, unable to form new short term memories, wouldn’t get any. ‘When you’ve reached that state, it is probably incurable; you have had so much damage to your hippocampus and other parts of the brain I can’t see that really coming back,’ says Doig. ‘And that’s what we’ve been testing drugs on.’
You need to take out the amyloid in people with preclinical Alzheimer’s, well before symptoms have appeared
The general consensus for the failure of all drugs tested so far for Alzheimer’s disease is that by the time cognitive symptoms appear, their brain has already accumulated decades of damage. As drug trials have shown, even treating people with the first mild cognitive symptoms using amyloid-targeting treatments hasn’t worked. ‘If you use a drug that targets the underlying cause, on a person with Alzheimer’s that has had this going on in their brain for 20 years, it is too little, too late,’ Martins says. ‘You need to take out the amyloid in people with preclinical Alzheimer’s, well before symptoms have appeared, while their brain is still relatively intact in the key regions.’
To test this theory, how do we identify people who are going to develop Alzheimer’s a decade or more before they first show any outward symptoms? That’s where the familial cases, with their very predictable age of onset, comes in. A person who inherits the faulty gene will definitely develop the disease, and symptoms will strike very close to the age their parent got it, says Anne Fagan from Washington University, who runs the biomarker analysis arm of the DIAN clinical trial. Among familial cases, researchers talk about estimated years to symptom onset (EYO). ‘With the combination of biomarkers and EYO we can pinpoint where somebody is along the disease process, and truly and properly evaluate the drug of interest,’ she says
The DIAN trial will test two experimental monoclonal antibody therapies, Roche’s gantenerumab and Eli Lilly’s solanezumab, both of which have failed big Phase III clinical trials in patients that already display mild cognitive symptoms. The trial recently completed patient recruitment. ‘Everybody in the trial has come in for their first evaluation,’ says Fagan. ‘It won’t be completely finished for a few years but some interim analyses are being performed.’
The results are eagerly anticipated. A successful trial would be fantastic news for people with the inherited form of the disease. But will the results translate to the other 99% of cases; will early intervention work there too? ‘That’s the $64,000 question,’ says Anne. ‘We don’t know, that will be the next step.’
Scanning the horizon
Other Alzheimer’s researchers are not waiting for the result of the DIAN study to answer that question. They’re looking to exploit another relatively recent development in the field – brain scans that can detect amyloid accumulation in the brains of living people.
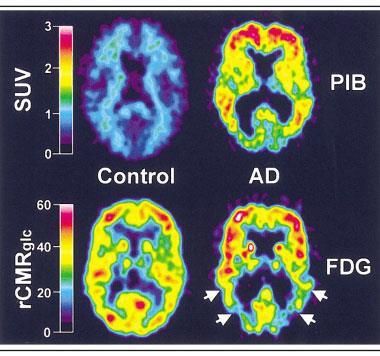
In the 2000s, researchers William Klunk and Chester Mathis at the University of Pittsburgh in the US took a molecule used as a histological dye for amyloid, thioflavin T, and added a radioactive carbon-11 atom to it. The resulting compound, Pittsburgh Compound B (PiB) – and the amyloid plaques it sticks to – is visible to positron emission tomography (PET) brain scans.8
‘The ability to look inside the brain and see these deposits during life was revolutionary,’ says Martins. ‘We could now identify people with Alzheimer’s with absolute certainty.’ The downside of the compound was carbon-11’s very short half-life: each time a batch of PiB was prepared, Martins says, you could scan two patients – if you were lucky. ‘Today we have about five different ligands, all fluorine-18. They have a longer half-life so we can look at eight or 10 people in a day.’
Brain imaging is now being used to enrol people for clinical trials aged over 65 years who show no cognitive symptoms of the disease, but for whom brain scans reveal are beginning to accumulate amyloid and are at high risk of developing the disease. The EARLY trial, started in 2015, is testing Janssen’s BACE 1 inhibitor in the preclinical Alzheimer’s population.9 The A4 trial, started in 2014, is using Lilly’s solanezumab antibody.
If that study fails, I think the amyloid story will be rapidly closed down for Alzheimer’s
Given the typical decade or more delay between amyloid accumulation and disease onset, answers from these trials might not be quickly forthcoming. The EARLY trial is not scheduled to end until 2024. Leaders of the A4 trial recently announced they were extending the trial so that participants in the double-blind study will be treated with an escalating dose of the drug (or a placebo) for four and a half years, rather than the three years originally planned.10
But the results will be worth waiting for, says Martins. We know these drugs can bring amyloid levels down, he says. ‘If that study fails, I think the amyloid story will be rapidly closed down for Alzheimer’s,’ he says. ‘If it’s going to work, this is going to be the study.’
James Mitchell Crow is a science writer based in Melbourne, Australia
NOTE: Article updated 20 June 2018 to correct the spelling of verubecestat
References
1 M A Lalli et al, Alzheimer’s & Dementia, 2014, 10, S277 (DOI: 10.1016/j.jalz.2013.09.005)
2 F Lopera et al, JAMA, 1997, 277, 793 (DOI: 10.1001/jama.1997.03540340027028)
3 R Yan, Transl Neurodegener., 2016, 5, 13 (DOI: 10.1186/s40035-016-0061-5)
4 C L Masters et al, EMBO J, 1985, 4, 2757 (https://bit.ly/2Ko2q8d)
5 R Yan, Transl. Neurodegener., 2016, 5, 13, (DOI: 10.1186/s40035-016-0061-5)
6 https://www.chemistryworld.com/news/dementia-slips-down-pharma-agenda/3008525.article
7 A J Doig et al, ACS Chem. Neurosci., 2017, 8, 1435 (DOI: 10.1021/acschemneuro.7b00188)
8 W E Klunk et al, Ann. Neurol., 2004, 55, 306 (DOI: 10.1002/ana.20009)
9 https://www.earlytrial.com/about-this-trial
11 S Ayton et al, Brain, 2017, 140, 2112 (DOI: 10.1093/brain/awx137)
12 B R Stockwell et al, Cell, 2017, 171, 273 (DOI: 10.1016/j.cell.2017.09.021)










No comments yet