



‘The internet is the only comparable network in existence,’ says Bartosz Grzybowski from the Ulsan National Institute of Science and Technology, South Korea, and the Polish Academy of Sciences. He is talking about Chematica – a computer network mapping millions of molecules and reactions in the known chemical universe. Organic synthetic chemists have questioned whether Chematica could herald a new era in synthesis planning, but Grzybowski is certain that such software will be an indispensable tool for making molecules.
‘Could the computer come up with a synthesis that would work, that was efficient?’ asks Varinder Aggarwal, an expert in organic synthesis at the University of Bristol, UK. ‘As soon as it gets close to doing anything like that, that’s a huge breakthrough.’
Making a complex product, be it a blockbuster drug or a DNA base, from simple building blocks is a battle. Researchers often have to recall reactions or mechanisms from memory and consult the literature to map out a synthesis. Even then, there are no guarantees it will work, the path often littered with low yields, escalating costs and non-existent chemicals.
A researcher’s memory is a key asset in synthesis planning, but it’s vulnerable. ‘After a year of not using a particular reaction, you keep forgetting the Pictet–Spengler cyclization, whatever it is,’ comments Grzybowski. ‘This knowledge evaporates with time.’
Chemical orienteering
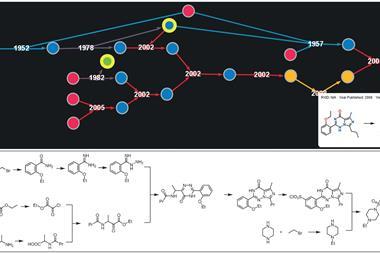
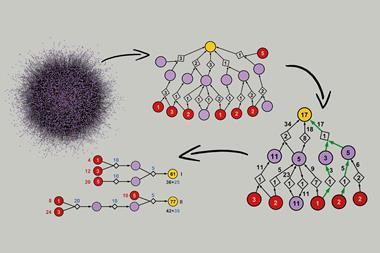
Chematica was born out of this frustration. Over 15 years, Grzybowski and his colleagues have manually input information on around 10 million substances and the reactions that link them together into a complex piece of software.1 ‘It’s really like an airport network,’ he explains. ‘You have hubs, you have connections.’
This network layout allows Chematica’s algorithms to search for pathways quickly, and in a fraction of the time it would take to sift through a database or list. It’s similar to how humans plot out a journey – mapping out a route on a London tube map, for instance, is far easier than trawling through a list of all station connections.
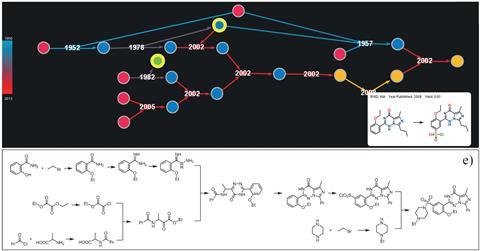
A chemist can input their destination (target molecule) into Chematica and search for any number of train lines (pathways) based on cost, substrate popularity or availability and number of steps – all in a matter of seconds. Each step, and the product it makes, is given a score based on two equations: the reaction and chemical scoring functions. The reaction scoring function (RSF) will penalise a move if the chemistry involved is difficult to carry out. But the chemical scoring function (CSF) will ascribe a score based on the molecule’s simplicity and whether it’s a known structure – the higher the CSF score, the more attractive the pathway.
These scoring functions allow Chematica to investigate different pathways, scrap them if they yield undesirable results, and move onto the next possible route. The longer the route extends, however, the greater the number of possibilities. Within five steps of the target molecule, the number of possible pathways can grow up to the order of 1020.
Every move you make
There’s a temptation to liken such intense computational acrobatics to the way in which computers have bested humans at chess or go – but it’s a flawed analogy, according to Grzybowski. ‘If you play chess, once you’re in a certain position with a certain arrangement of pieces on the board, you don’t look back,’ he says. ‘You’re just looking forward.’
Chemistry presents an altogether larger problem and Grzybowski likens synthetic planning to a combination of chess and a Rubik’s cube: ‘The position is how many steps you took and whether these steps were required because not every step is equal to each other.’But what if you want to plan a synthesis for a product that can’t be found in the literature – a complete unknown? Such calculations require good quality data and basing a pathway on what has gone before is probably a step too far, according to Richard Whitby, who led the ‘Dial-a-molecule’ network, a scheme set up to assess automation in chemistry.
Past retrosynthetic efforts such as ARChem and SynChem have suffered because the software automatically draws up chemical rules from Reaxys– a database of known chemical literature. ‘The data is full of errors and inadequacies,’ says Whitby. ‘Those get carried forward so that a lot of the suggestions that ARChem and SynChem come up with are just plain stupid.’
‘People started thinking machine learning will do it: “I go to Reaxys and I automatically extract the reaction rules somehow”,’ laments Grzybowski. ‘You must teach the computer much more – you must teach it reactivity conflicts, protection chemistries, which groups are incompatible with a reaction type and so on.’
Whitby goes on to explain that another program, Logic and Heuristics Applied to Synthetic Analysis, developed by E J Corey in the 1970s, took a different approach.2 ‘The rules … were written by organic chemists with their knowledge of chemistry,’ he says.
Although LHASA was a significant moment, it was limited in what it could achieve. The sheer number of rules required to solve the problem outweighed the number Corey’s team could manually input, according to Grzybowski. ‘Corey had 300, not 30,000, [rules] and nobody wants to code by hand,’ he explains. It’s why some opted for the machine learning route in the first place.
Bull by the horns
But Grzybowski has revisited this idea of moulding an automated platform through manual work to set another bull loose in the china shop: Syntaurus.3 Over the past decade, Grzybowski’s group has manually encoded over 20,000 chemical rules into the software. These reaction rules account for incompatible groups, protection chemistries and even small differences in bond angle and length.
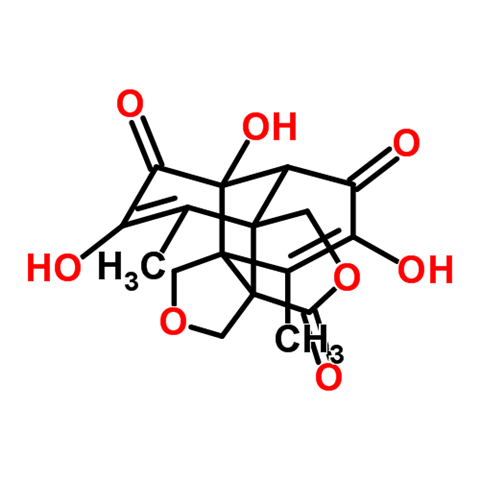
Given the team couldn’t rely on machine learning to produce Syntaurus, it made for an intimidating challenge. ‘You realise you either do it the hard way or you don’t do it at all,’ says Grzybowski. ‘By now this entire thing, the rule base for chemistry, is much larger than, I think, Encyclopaedia Britannica.’
In their most recent piece of research, Grzybowski’s group has demonstrated Syntaurus’s potential. The program has mapped out a total synthesis for epicolactone, a complex natural product isolated from a plant fungus in 2012. Synthetic chemists have taken nearly three years to design a plausible biosynthetic pathway for the compound. Syntaurus computed a similar pathway in a couple of hours.
Making converts
For all these demonstrations, however, there remains a healthy amount of scepticism about Chematica and Syntaurus. ‘I think the next step is to get the computer to come up with a synthesis of a relatively complex molecule and then to demonstrate it in the lab,’ says Aggarwal. ‘If you can do that on a relatively complex molecule … and be honest about it, then I think you’ve got something.’
For Whitby, seeing it in action will be the ultimate test. ‘When and if he actually produces Chematica, it will probably give good, sensible results,’ he says. ‘We’d all be much, much more convinced if we could actually use it.’
But Grzybowski claims that Syntaurus has already taken on Aggarwal’s challenge – and succeeded. Although he’s tight-lipped on the subject, his team say that they mapped out the shortest and cheapest synthesis for a blockbuster drug and successfully performed it in the lab.
The true value of Syntaurus remains to be seen, but harnessing computer memory to preserve our own and promote new chemistry may well be the way forward. ‘If someone’s designing a new drug or material, you can work out what molecule you really want … and then a chemist can make it in a few weeks,’ says Whitby. ‘When it works well, it will be transformative.’
Correction: On 10 August a quote was updated owing to a technical error in it
References
1 M Kowalik et al, Angew. Chem. Int. Ed., 2012, 51, 7928 (DOI: 10.1002/anie.201202209)
2 E J Corey et al, JACS, 1972, 94, 421 (DOI: 10.1021/ja00757a020)
3 S Szymkuć et al, Angew. Chem. Int. Ed., 2016, 55, 5904 (DOI: 10.1002/anie.201506101)













No comments yet